
Zusammenfassung
Das Gehirn ist reich an Arachidonsäure (ARA) und Docosahexaensäure (DHA), langkettigen mehrfach ungesättigten Fettsäuren (LCPUFAs) der n-6- bzw. n-3-Reihe. Beide sind essentiell für die optimale Entwicklung und Funktion des Gehirns. Eine diätetische Anreicherung mit DHA und anderen langkettigen n-3 PUFA, wie Eicosapentaensäure (EPA), zeigte positive Effekte auf Lernen und Gedächtnis, neuroinflammatorische Prozesse sowie synaptische Plastizität und Neurogenese. ARA, DHA und EPA sind Vorläufer einer vielfältigen Reihe bioaktiver Lipidmediatoren, einschließlich Endocannabinoide. Das Endocannabinoid-System umfasst Cannabinoid-Rezeptoren, deren endogene Liganden, die Endocannabinoide, sowie deren Biosynthese- und Abbauenzyme. Anandamid (AEA) und 2-Arachidonoylglycerol (2-AG) sind die am besten untersuchten Endocannabinoide und leiten sich beide von Phospholipid-gebundener ARA ab. Das Endocannabinoid-System spielt ebenfalls eine etablierte Rolle bei Neuroinflammation, synaptischer Plastizität und Neurogenese, was auf eine Überschneidung der neuroprotektiven Effekte dieser verschiedenen Lipidklassen hindeutet. Tatsächlich deutet eine wachsende Evidenz auf ein komplexes Zusammenspiel zwischen n-3- und n-6-LCPUFA und dem Endocannabinoid-System hin. Zum Beispiel reduziert eine langfristige DHA- und EPA-Supplementierung die AEA- und 2-AG-Spiegel, mit reziproken Anstiegen der Spiegel analoger endocannabinoidähnlicher, DHA- und EPA-abgeleiteter Moleküle. Diese Übersicht fasst die aktuelle Evidenz dieses Zusammenspiels zusammen und erörtert das therapeutische Potenzial für Gehirnschutz und -reparatur.
Schlüsselwörter: Endocannabinoid-System, Neurogenese, Neuroinflammation, Omega-3-Fettsäuren, Omega-6-Fettsäuren
Einleitung
N-6- und n-3-langkettige mehrfach ungesättigte Fettsäuren (LCPUFA) sind essentielle Bestandteile von Membranphospholipiden und zudem Vorläufer eines großen und stetig wachsenden Repertoires bioaktiver Lipidmediatoren. Das Gehirn ist besonders reich an der n-6 PUFA Arachidonsäure (ARA) und der n-3 PUFA Docosahexaensäure (DHA), wobei beide für eine optimale Gehirnentwicklung und -funktion unerlässlich sind [1]. Eine erhöhte diätetische Zufuhr von DHA und Eicosapentaensäure (EPA), einer weiteren n-3 LCPUFA, wirkt sich positiv auf Lernen und Gedächtnis aus, reduziert neuroinflammatorische Prozesse und fördert die synaptische Plastizität und Neurogenese [2]. Ähnlich zeigen sich typischerweise inverse Beziehungen zwischen Fischkonsum oder DHA-Blutspiegeln und altersbedingtem kognitivem Verfall [3]. Aktuelle Schätzungen weisen jedoch darauf hin, dass viele Bevölkerungsgruppen weltweit DHA und EPA in Mengen konsumieren, die weit unter den Empfehlungen internationaler Behörden liegen [4–6]. Eine ausgewogene Zufuhr dieser wichtigen Fettsäuren ist entscheidend für ein Immunsystem stärken und die allgemeine Gesundheit.
Der Wirkungsmechanismus der LCPUFA ist noch unzureichend verstanden und wird durch das vielfältige Repertoire an bioaktiven Lipidmediatoren, die erzeugt werden können, zusätzlich erschwert. So ist beispielsweise ARA der Vorläufer einer breiten Palette von Mediatoren, darunter die beiden wichtigsten Endocannabinoide im Gehirn [7]. Dem Endocannabinoid-System wurden ebenfalls wichtige Rollen bei neuroprotektiven und pro-neurogenen Prozessen zugeschrieben, wie der Abschwächung chronischer Neuroinflammation, der Regulierung der pro-inflammatorischen Zytokin-Freisetzung und der Förderung der synaptischen Plastizität und der adulten Neurogenese [8, 9]. Wichtig ist, dass es therapeutisches Potenzial bei Gehirnalterung und neurodegenerativen Erkrankungen gezeigt hat [10].
Es gibt also eine beträchtliche Überschneidung in den Effekten von n-3 PUFA und dem Endocannabinoid-System; jedoch wurden diese unterschiedlichen Klassen von Lipidmediatoren traditionell getrennt betrachtet und erforscht. Diese Sichtweise wird nun in Frage gestellt, da eine wachsende Anzahl unabhängiger Beweise auf ein komplexes Zusammenspiel zwischen ihnen hindeutet. Beispielsweise wurden analoge Serien von Ethanolamid-Endocannabinoid-ähnlichen Molekülen, die von DHA und EPA abgeleitet sind, identifiziert, obwohl ihre biologischen Rollen noch nicht etabliert sind [11, 12]. Des Weiteren zeigte eine aktuelle elegante Arbeit, dass ein langfristiger diätetischer Mangel an n-3 PUFA bei Mäusen endocannabinoid-vermittelte neuronale Funktionen in verschiedenen Gehirnregionen aufhob. Dies zeigte erstmals, wie das Endocannabinoid-System durch Manipulation des diätetischen n-6:n-3 PUFA-Verhältnisses reguliert werden kann [13–15]. Dies ist besorgniserregend, da die westliche Ernährung typischerweise ein n-6:n-3 PUFA-Verhältnis von etwa 15:1 aufweist, während das ideale Verhältnis näher an 4:1 liegen sollte [16]. Diese unausgewogene Aufnahme spiegelt sich in niedrigen bis sehr niedrigen Gewebespiegeln von DHA und EPA wider [17] und könnte auch an der Ätiologie vieler Krankheiten wie Herz-Kreislauf-Erkrankungen, Krebs, entzündlichen und Autoimmunerkrankungen beteiligt sein [16].
Das Ziel dieser Übersicht ist es, die aktuelle Evidenz des Zusammenspiels zwischen n-3- und n-6-LCPUFA und dem Endocannabinoid-System zusammenzufassen und die potenzielle Rolle der Modifikation ihrer Spiegel durch diätetische Manipulation der n-6- und n-3-PUFA-Zufuhr zu diskutieren, mit dem Ziel, Neuroinflammation zu lindern und Gehirnschutz und -reparatur, insbesondere im Alter, zu fördern.
Stoffwechsel von PUFA und Endocannabinoiden
ARA und DHA sind die beiden Haupt-PUFA im Gehirn [2]. Diese LCPUFA können entweder in vorgebildeter Form über die Nahrung zugeführt oder in der Leber aus ihren kürzerkettigen Vorläufern, Linolsäure (LA, 18:2n-6) und α-Linolensäure (ALA, 18:3n-3), synthetisiert werden [18, 19]. Die Effizienz der Umwandlung beim Menschen ist jedoch extrem begrenzt [20], und aufgrund der gemeinsamen Natur der biosynthetischen Wege führt ein Ungleichgewicht in der diätetischen Aufnahme von LA und ALA zu einer wechselseitigen Hemmung des jeweiligen anderen Weges und schränkt die Umwandlung weiter ein [21]. Daher ist der effizienteste Weg zur Erhöhung der Gewebespiegel von LCPUFA die Aufnahme der vorgebildeten LCPUFA. Die Biosynthesewege von n-6 und n-3 PUFA sind in Fig. 1 detailliert dargestellt.
Fig. 1.
 Stoffwechselwege von n-6 und n-3 PUFA sowie Lipidmediatoren aus ARA, DHA und EPA im GehirnOpen in a new tab
Stoffwechselwege von n-6 und n-3 PUFA sowie Lipidmediatoren aus ARA, DHA und EPA im GehirnOpen in a new tab
Die Synthese von n-6 und n-3 LCPUFA beginnt mit der Desaturierung von LA und ALA zu γ-Linolensäure (GLA, 18:3n-6) bzw. Stearidonsäure (18:4n-4), katalysiert durch Δ6-Desaturase (FADS2-Gen). GLA wird zu Dihomo-γ-Linolensäure (DGLA, 20:3n-6) und SDA zu Eicosatetraensäure (20:4n-3) verlängert (ELOVL1-Gen). Die Δ5-Desaturase (FADS1-Gen) wandelt DGLA in ARA (20:4n-6) und 20:4n-3 in EPA (Timnodonsäure, 20:5n-3) um. Zwei Elongationszyklen (Elongase-2, ELOVL2-Gen) wandeln ARA in Adrensäure (AdA, 22:4n-6) und dann Tetracosatetraensäure (24:4n-6) um, und EPA in Docosapentaensäure (DPAn-3, Clupanodonsäure, 22:5n-3) und dann Tetracosapentaensäure (24:5n-3). Eine zweite Desaturierung durch Δ6-Desaturase produziert Tetracosapentaensäure (24:5n-6) bzw. Tetracosahexaensäure (Nisinsäure, 24:6n-3). Diese werden zur β-Oxidation durch Acyl-Coenzym-A-Oxidase (ACOX1-Gen) und d-bifunktionelles Enzym (HSD1784-Gen) sowie peroxisomale Thiolasen in das Peroxisom transportiert, um Docosapentaensäure (DPAn-6, Osbonsäure, 22:5n-6) und DHA (Cervonsäure, 22:6n-3) zu produzieren, die dann zurück in das Endoplasmatische Retikulum transportiert werden.
Die endogene Synthese von LCPUFA ist im Gehirn im Vergleich zur Aufnahme aus dem unveresterten Plasma-Fettsäurepool gering [22, 23], was darauf hindeutet, dass die Gehirnspiegel durch Aufnahme aus diätetischen und/oder Leberquellen im Blut aufrechterhalten werden. Obwohl LCPUFAs die Blut-Hirn-Schranke über einfache Diffusion zu überwinden scheinen [24], wurden aktive Transporter identifiziert, die eine Rolle bei der Regulierung der Spezifität von LCPUFA-Konzentrationen spielen könnten [20]. Zudem wurden mehrere Mechanismen, darunter β-Oxidation, verringerte Inkorporation, Elongation und geringerer Phospholipid-Recycling, identifiziert, die die hohe ARA- und DHA-Konzentration im Verhältnis zu anderen LCPUFAs aufrechterhalten [25, 26]. Die LCPUFA-Zusammensetzung des Gehirns reagiert jedoch auf die Nahrungsaufnahme, sodass eine LA-reiche Ernährung mit einem LA:ALA-Verhältnis von 10:1, typisch für eine westliche Ernährung, die DHA-Anreicherung im Gehirn verringert und die Spiegel von Adrensäure (AdA, Docosatetraensäure, 22:4n-6) und Docosapentaensäure (DPAn-6, 22:5n-6) erhöht [27]. Eine Ernährung mit einem LA:ALA-Verhältnis von 1:1, die unserer evolutionären Entwicklung ähnlicher ist [16], führt zu höheren DHA-Spiegeln im Gehirn. Ungleichgewichte in der Aufnahme beeinträchtigen nicht nur den LCPUFA-Gehalt des Gehirns, sondern können auch die Produktion einer breiten Palette von Mediatoren beeinflussen, die aus diesen LCPUFA stammen, wodurch potenziell die Gehirnaktivität und -funktion negativ beeinflusst werden.
Die Fettsäurezusammensetzung neuronaler Membranen beeinflusst die Zellfunktion durch direkte Effekte auf die biophysikalischen Eigenschaften der Membran, aber auch durch die Bereitstellung eines Vorläuferpools für Signalmoleküle und lipidabgeleitete Mediatoren [1]. N-6- und n-3-LCPUFA sind die Vorläufer einer Vielzahl bioaktiver Mediatoren, die an vielen zellulären Prozessen beteiligt sind, insbesondere im Zusammenhang mit der Entzündungsreaktion [2]. Drei Hauptwege sind an der Produktion dieser Oxylipin-Mediatoren beteiligt: (1) Cyclooxygenase (COX, auch bekannt als Prostaglandin-Endoperoxid-H-Synthase oder PGHS) und nachfolgende Synthasen, (2) Lipoxygenase (LOX) und (3) Cytochrom P450-Mischfunktion-Oxidase-Enzyme (CYP) [28]. Diese kanonischen Wege produzieren die klassischen Mediatoren, wobei diejenigen, die aus C20 PUFA wie ARA und EPA stammen, Eicosanoide genannt werden, während diejenigen aus C22 PUFA wie DHA Docosanoide genannt werden. Analoge Serien von Oxylipinen, die aus LA, Dihomo-γ-Linolensäure (DGLA), AdA und ALA sowie der n-3 Docosapentaensäure (DPAn-3) erzeugt werden, wurden ebenfalls identifiziert, aber ihre Rollen sind in der Literatur nicht gut charakterisiert und daher nicht der Fokus dieser Übersicht. Interessierte Leser werden jedoch auf eine exzellente Übersicht von Gabbs und Kollegen verwiesen [29].
COX katalysiert die anfängliche Oxygenierung nicht-veresterter Fettsäuren zur Produktion von Prostaglandin H (PGH), einem kurzlebigen Zwischenprodukt, das weiter zu Prostanoiden metabolisiert wird, wie z.B. anderen Prostaglandin-Serien (PGD, PGE, PGF), Prostacyclinen (PGI), Thromboxanen (Tx) und Lipoxinen (Lx), Hydroxy- und Hydroperoxy-Fettsäuren [30]. Wirbeltiere haben zwei Hauptisoformen von COX: COX-1 und COX-2 [31]. COX-1 wird konstitutiv exprimiert, während COX-2, obwohl ein induzierbares Enzym in den meisten Geweben, im Kortex, Hippocampus und der Amygdala konstitutiv exprimiert wird [32, 33]. COX-2 ist nicht nur ein Schlüsselenzym in entzündlichen und neuroinflammatorischen Prozessen, sondern spielt auch eine wichtige Rolle bei der Regulierung neuronaler Aktivität, wie Lernen und Gedächtnis [34]. COX-2 oxygeniert eine breite Palette von Fettsäuren und Fettsäureestern [35].
COX-2 galt traditionell als verantwortlich für die Verursachung von Entzündungen und Neuroinflammation durch die Umwandlung von ARA in PG und Tx; dieses vereinfachte Modell wurde jedoch mit einem besseren Verständnis des empfindlichen Gleichgewichts zwischen positiven und negativen Rückkopplungsschleifen überdacht [36]. Beispielsweise sind PGE2 und PGD2 pro-inflammatorische Mediatoren, die für die Induktion von Entzündungen verantwortlich sind, aber in einem späteren Stadium des Prozesses auch für den Klassenwechsel der Eicosanoidproduktion von PG und Leukotrienen (Lt) zu Lx verantwortlich sind [36]. Es wurde konsistent gezeigt, dass eine Erhöhung der diätetischen n-3 PUFA das Lipidprofil von Membranen verändert und das Gleichgewicht von n-6- und n-3-PUFA, die als Substrate für COX konkurrieren, beeinflusst. Dies verändert folglich die synthetisierten Prostaglandin-Serien, was letztendlich die zellulären Reaktionen auf mitogene und entzündliche Stimuli verändert [37–41]. Dies wurde in vielen Zellen im ganzen Körper, einschließlich Gliazellen, nachgewiesen [42].
LOX katalysieren die Bildung von Hydroxylfettsäuren und deren Metaboliten wie Lt, Lx und den “spezialisierten Lipidmediatoren” (SPM) [29]. Dazu gehören die Resolvine (Rv), Protectine (PD) und Maresine (MaR), die von n-3 LCPUFA stammen [43]. LOX-Enzyme werden traditionell nach der Position der Hydroxyl- und Hydroperoxy-Fettsäuren klassifiziert, die sie aus ARA produzieren, z. B. bildet 5-LOX 5-Hydroxy-Eicosatetraensäure (5-HETE) und 5-Hydroperoxy-Eicosateraensäure (5-HpETE); dieses System hat jedoch Einschränkungen, da die Position je nach Kettenlänge der Substrate variiert und einige LOX an mehr als einer Position wirken [29].
Die SPMs sind eine schnell wachsende Klasse von Molekülen, die an der aktiven Auflösung von Entzündungen beteiligt sind und über COX- und LOX-katalysierte Wege produziert werden [43]. D-Serien-Resolvine (RvD), PD und MaR werden aus DHA produziert, während E-Serien-Resolvine (RvE) aus EPA stammen [44]. Eine weitere Serie von RvD und MaR wurde kürzlich identifiziert, die aus DPAn-3 generiert werden, darunter RvD1n-3 DPA und MaR1n-3 DPA, welche ähnliche entzündungshemmende und pro-resolvierende Eigenschaften wie die von DHA und EPA aufweisen [45, 46]. Die SPMs wirken über eine Reihe von zellspezifischen Rezeptoren, zum Beispiel bindet RvD1 an GPR32 und den Lipoxin-A4-Rezeptor (ALx), und RvE1 bindet an den ChemR23-Orphan-Rezeptor und den Leukotrien-B4-Rezeptor (BLT1) [47]. Das am besten charakterisierte SPM hinsichtlich des Nervensystemschutzes ist (Neuro)protectin D1 (NPD1, 10R-17S-Dihydroxy-Docosahexaensäure), das als Reaktion auf Verletzungen biosynthetisiert wird und therapeutisches Potenzial bei einer Vielzahl neurologischer Erkrankungen haben könnte [48, 49]. Darüber hinaus blockiert die Acetylierung von COX-2 durch Aspirin die PG-Biosynthese, aber COX-2 ist immer noch in der Lage, HETE aus ARA, Hydroxy-Docosahexaensäure (HDoHE) aus DHA und Hydroxy-Eicosapentaensäure (HEPE) aus EPA zu produzieren, die von Leukozyten in Aspirin-getriggerte Formen von Lx, Rv und PD umgewandelt werden können [50].
Eine weitere Klasse von Metaboliten, die aus n-3 PUFA durch LOX erzeugt werden, sind die elektrophilen Fettsäure-Oxo-Derivate (EFOX), wobei 7-oxo-DHA, 7-oxo-DPA und 5-oxo-EPA aus DHA, DPAn-3 bzw. EPA produziert werden [51, 52]. EFOX zeigen eine breite Palette entzündungshemmender Wirkungen, einschließlich der Funktion als Agonisten nukleärer Rezeptoren wie dem Peroxisom-Proliferator-aktivierten Rezeptor (PPAR) und der Hemmung der Zytokinproduktion in aktivierten Makrophagen [52]. Darüber hinaus erhöht die Acetylierung von COX-2 durch Aspirin, im Einklang mit der Bildung von Aspirin-getriggerten SPM, die Bildung von EFOX signifikant [2].
Der dritte oxidative Weg beinhaltet CYP-Epoxygenasen und ϖ-Hydrolasen, die PUFA zu Lipidmediatoren mit vielen unterschiedlichen biologischen Funktionen auf systemischer und zellulärer Ebene metabolisieren [53, 54]. Regio- und Stereoisomere von Epoxy-Eicosatetraensäuren (EET) und HETE werden aus ARA produziert, während die aus EPA abgeleiteten Epoxy-Eicosatetraensäuren (EETeTR) und Hydroxy-Eicosapentaensäuren (HEPE) und Epoxy-Docosapentaensäuren (EDP) sowie HDoHE aus DHA stammen [54]. EPA ist das bevorzugte Substrat für die meisten CYP-Isoformen, wobei der Metabolismus von DHA und ARA mit ähnlichen Raten erfolgt [54]. Die Expression von CYP-Isoformen erfolgt in mehreren Zelltypen im gesamten Gehirn, einschließlich Astrozyten, Neuronen und Endothelzellen [53].
Darüber hinaus sind n-6- und n-3-PUFAs auch Vorläufer endogener Liganden der Endocannabinoid-Rezeptoren (Endocannabinoide). Das Endocannabinoid-System besteht aus den Cannabinoid-Rezeptoren (CB1- und CB2-Rezeptoren), Endocannabinoiden und den für die Endocannabinoid-Synthese und -Abbau erforderlichen Enzymen [55]. Zwei Familien von Endocannabinoiden wurden identifiziert: 2-Acylglycerole und Ethanolamide; jedoch sind nicht alle Konjunktionsprodukte Liganden der Cannabinoid-Rezeptoren [56]. Die am häufigsten vorkommenden und am besten charakterisierten Endocannabinoide im Gehirn sind das 2-Acylglycerol 2-Arachidonoylglycerol (2-AG) und das Ethanolamid N-Arachidonoylethanolamin (AEA, Anandamid), die beide aus ARA abgeleitet sind [7]. Weitere von n-6 PUFA abgeleitete Endocannabinoide sind Dihomo-γ-Linolenoyl-Ethanolamid, Docosatetraenoyl-Ethanolamid, 2-Arachidonylglycerolether (Noladin-Ether), O-Arachidonoylethanolamin (Virodhamin) und N-Arachidonoyldopamin; obwohl diese Endocannabinoide an Cannabinoid-Rezeptoren binden können, ist ihre Funktion noch unklar und wird daher in dieser Übersicht nicht weiter diskutiert [57]. Analoge Serien von Endocannabinoiden wurden aus n-3 PUFA identifiziert. Alpha-Linolenoylethanolamid (ALEA) wird aus ALA produziert und wurde im menschlichen Plasma identifiziert, wo die Spiegel nach ALA-Supplementierung diätetisch beeinflussbar waren [58]. Die am besten charakterisierten von n-3 PUFA abgeleiteten Endocannabinoide werden jedoch aus DHA und EPA produziert, wobei die 2-Acylglycerole 2-Docosahexaenoylglycerol (2-DHG) und 2-Eicosapentaenoylglycerol (EPG) sowie die Ethanolamide N-Docosahexaenoylethanolamin (DHEA) und N-Eicosapentaenoylethanolamin (EPEA) jeweils aus DHA und EPA generiert werden [12, 59]. Diese Übersicht wird sich auf die von ARA, DHA und EPA abgeleiteten Endocannabinoide konzentrieren.
AEA und 2-AG werden aus membrangebundener Phospholipid-ARA produziert, wobei die Synthese am postsynaptischen Terminal über erhöhte intrazelluläre Kalziumspiegel erfolgt. Beide werden bedarfsgesteuert gebildet und schnell zu ARA abgebaut oder zu weiteren bioaktiven Mediatoren oxygeniert [60]. Die Hauptwege für die Biosynthese und den Abbau von AEA und 2-AG werden unten beschrieben und in Fig. 3 zusammengefasst. Die genaue Natur dieser Wege muss jedoch noch geklärt werden, da die Komplexität des Endocannabinoid-Systems und die Existenz multipler, oft redundanter Wege dies erschweren [61].
Fig. 3.
Open in a new tab
Zusammenspiel bei der Synthese und den Wirkungen der 2-Acylglycerole und Ethanolamide, die von ARA, DHA und EPA abgeleitet sind. Der Hauptweg für die AEA-Produktion beginnt mit der N-Acyltransferase (NAT), die ARA von Phosphatidylcholin (ARA-PC) auf Phosphatidylethanolamin (PE) überträgt, um N-Arachidonoyl-Phosphatidylethanolamin (NArPE) zu erzeugen. Es folgt die Hydrolyse durch eine N-Acyl-Phosphatidylethanolamin-selektive Phospholipase D (NAPE-PLD) zur Produktion von AEA. Weitere Wege umfassen die NAPE-Deacylierung durch die α/β-Hydrolase-Domäne 4 (ABHD4) und entweder die produzierte Glycerophosphoarachidonoylethanolamid (GP-NAPE), die durch Phosphodiesterase (PDE) zu AEA gespalten wird, oder Lyso-NAPE, die durch Lyso-NAPE-Phospholipase D (PLD) direkt zu AEA hydrolysiert wird. NAPE kann auch durch Phospholipase C (NAPE-PLC) hydrolysiert werden, um Phospho-Anandamid (PAEA) zu erzeugen, das dann durch Phosphatasen wie die Protein-Tyrosin-Phosphatase (PTPN22) zu AEA dephosphoryliert wird. Die Produktion von DHEA und EPEA aus Phospholipid-gebundenem DHA und EPA scheint dieselben Wege zu teilen. Die Synthese von 2-AG erfolgt aus Phosphatidylinositol-gebundener ARA (ARA-PI) über Phospholipase C-β (PLCβ) und die Produktion eines ARA-Diacylglycerols (DAG), das durch Diacylglycerol-Lipasen-α zu 2-AG hydrolysiert wird. Weitere Wege umfassen die Dephosphorylierung von 2-AG-Lysophosphatidsäure (2-AG-LPA) durch eine LPA-Phosphatase (2-LPA-P) oder über die sequentielle Wirkung von PLA1, die PI in 2-Arachidonoyl-Lyso-PI (2-AG-LPI) und dann durch Lyso-Phospholipase C (Lyso-PLC) in 2-AG umwandelt. Die Wege der 2-DPG- und 2-EPG-Produktion sind derzeit unbekannt. 2-AG und AEA wirken an CB1- und CB2-Rezeptoren, GPR55 und PPAR, wobei AEA zusätzlich an TRPV-1 wirkt (grau dargestellt). Eine diätetische DHA- und EPA-Anreicherung reduziert Phospholipid-ARA und erhöht Phospholipid-DHA und EPA und begünstigt die Produktion von DHA- und EPA-abgeleiteten Endocannabinoiden, während eine akute DHA- und EPA-Behandlung in vitro 2-AG erhöht. DHA und EPA regulieren auch die Aktivität und Spiegel von CB1-, CB2-, TRPV-1- und PPAR-Rezeptoren. Detaillierte Erklärungen finden Sie im Text.
Die AEA-Produktion erfolgt über eine Reihe von Schritten aus dem membrangebundenen Phospholipid-Vorläufer sn-1 ARA-Phosphatidylcholin [62]. Eine kalziumabhängige N-Acyltransferase (NAT) überträgt ARA auf das Stickstoffatom von Phosphatidylethanolamin (PE), um N-Arachidonoyl-Phosphatidylethanolamin (NArPE) zu erzeugen, gefolgt von der Hydrolyse durch eine N-Acyl-Phosphatidylethanolamin-selektive Phospholipase D (NAPE-PLD) zur Produktion von AEA [63]. Weitere parallele Wege wurden identifiziert, wobei NAPE durch α/β-Hydrolase-Domäne 4 (ABHD4) deacyliert wird und entweder das produzierte Glycerophosphoarachidonoylethanolamid (GP-NAPE) durch eine metalldependente Phosphodiesterase (PDE) zu AEA gespalten wird oder Lyso-NAPE direkt durch Lyso-NAPE-Phospholipase D (PLD) zu AEA hydrolysiert wird. NAPE kann auch durch Phospholipase C (NAPE-PLC) hydrolysiert werden, um Phospho-Anandamid (PAEA) zu erzeugen, das durch Phosphatasen wie Protein-Tyrosin-Phosphatase 22 (PTPN22) zu AEA dephosphoryliert wird [63]. Studien mit NAPE-PLD-Knockout-Mäusen zeigen, dass NAPE-PLD der Hauptweg für die NAPE-Hydrolyse ist; die Bildung von AEA im Gehirn erfolgt jedoch auch leicht über NAPE-PLD-unabhängige Wege [64, 65].
Der Hauptweg für die Synthese von 2-AG im Gehirn erfolgt aus Phosphatidylinositol (PI)-gebundener ARA über Phospholipase C-β (PLCβ), die sn-1-Acyl-2-Arachidonoylglycerol, ein ARA-Diacylglycerol (DAG), produziert [66]. DAG wird dann durch die Wirkung von Diacylglycerol-Lipasen-α oder -β (DAGL-α oder DAGL-β) hydrolysiert, wobei die Acylgruppe entfernt wird [66]. DAGLα scheint die Hauptisoform für die 2-AG-Bildung im Gehirn zu sein, da der basale und stimulusinduzierte 2-AG-Gehalt des Gehirns bei DAGLα-, aber nicht bei DAGLβ-Knockout-Mäusen stark reduziert ist [67]. Weitere Wege zur Synthese von 2-AG umfassen die Dephosphorylierung von 2-AG-Lysophosphatidsäure (2-AG-LPA) durch eine LPA-Phosphatase (2-LPA-P) oder über die sequentielle Wirkung von PLA1, die PI in 2-Arachidonoyl-Lyso-PI (2-AG-LPI) und dann durch Lyso-Phospholipase C (Lyso-PLC) in 2-AG umwandelt [66].
DHEA und EPEA scheinen über dieselben Biosynthesewege wie AEA produziert zu werden [68], während die Synthese von 2-DHG und 2-EPG in der Literatur nicht gut charakterisiert ist. Es ist jedoch wahrscheinlich, dass sie über dieselben Wege wie 2-AG produziert werden, da eine chronische DHA- und EPA-Supplementierung die 2-AG- und AEA-Spiegel in verschiedenen Geweben, einschließlich des Gehirns, reduziert, mit reziproken Anstiegen der Spiegel von DHEA und 2-DHG sowie 2-EPG [12, 69–72]. Diese Veränderungen deuten auf eine Konkurrenz um gemeinsame Biosynthesewege hin, da DHA und EPA ARA aus Membranphospholipiden verdrängen. Interessanterweise zeigte eine kürzlich durchgeführte Arbeit in unserem Labor, dass eine akute Verabreichung von DHA oder EPA die 2-AG-Spiegel, jedoch nicht die AEA-Spiegel, in neuralen Stammzellen signifikant erhöhte [73]. Dieser Anstieg könnte durch die Konkurrenz um inaktivierende Enzyme wie COX-2 angetrieben werden, obwohl weitere Arbeiten erforderlich sind, um die zugrunde liegenden Mechanismen vollständig aufzuklären.
AEA und 2-AG wirken überwiegend an den G-Protein-gekoppelten Rezeptoren (GPCR) CB1 und CB2 [74]. Der CB1-Rezeptor ist im Gehirn weit verbreitet, wo er der häufigste GPCR ist und im Kortex, Hippocampus, Kleinhirn und den Basalganglien hoch exprimiert wird [74]. CB2-Rezeptoren wurden ursprünglich in Zellen des Immunsystems identifiziert [75], wurden aber in jüngerer Zeit auch in Glia und Untergruppen von Neuronen im Gehirn beschrieben [76]. Darüber hinaus wurde gezeigt, dass AEA und 2-AG auch an den Orphan-Rezeptor GPR55 [77] und die Peroxisom-Proliferator-aktivierten Rezeptoren (PPAR) [78] wirken. PPARs sind nukleär wirkende Transkriptionsfaktoren mit drei Subtypen, α, β (δ) und γ, und sind an vielen zellulären Prozessen beteiligt; zum Beispiel reguliert PPARγ Gene, die an neuroinflammatorischen Prozessen beteiligt sind [79]. AEA ist auch ein Ligand für den transienten Rezeptorpotenzial-Vanilloidrezeptor Typ 1 (TRPV-1), der in peripheren sensorischen Neuronen und im zentralen Nervensystem (ZNS) exprimiert wird, wo sie eine Rolle bei der Regulierung der synaptischen Funktion spielen [80].
Andere Endocannabinoide als 2-AG und AEA binden entweder nicht orthosterisch an CB1- oder CB2-Rezeptoren oder binden mit wesentlich geringerer Affinität; sie zeigen jedoch immer noch cannabimimetische Aktivitäten und potenzieren die Aktivität von 2-AG und AEA in einem Phänomen, das als „Entourage-Effekt“ bezeichnet wird [56, 81]. Es gibt jedoch Hinweise darauf, dass die Beziehung zwischen 2-AG und AEA und ihren Konjunktionsprodukten wesentlich nuancierter ist, und es wurde berichtet, dass andere Endocannabinoide entweder als funktionelle Antagonisten dienen [81] oder über nicht-endocannabinoidale Wege wirken. Zum Beispiel aktiviert DHEA Protein-Kinase-A (PKA)/cAMP-Response-Element-Binding-Protein (CREB)-Wege [82].
Über den Prozess des Endocannabinoid-Transports über Zellmembranen ist wenig bekannt, obwohl ein mutmaßlicher Endocannabinoid-Zellmembrantransporter an der Kontrolle des AEA- und 2-AG-Transports und -Metabolismus beteiligt ist [83]. Die Hydrolyse von AEA setzt ARA und Ethanolamin frei und wird hauptsächlich durch das Fatty Acid Amide Hydrolase (FAAH)-Enzym erreicht [84], obwohl weitere, noch zu identifizierende Proteine wahrscheinlich an dem Prozess beteiligt sind [61]. DHEA ist auch ein Substrat für die FAAH-Hydrolyse, um DHA und Ethanolamin freizusetzen [68], während der Prozess der EPEA-Hydrolyse noch identifiziert werden muss. Im Gegensatz zu den Ethanolamiden sind eine Vielzahl von Enzymen für den Abbau von 2-AG zu ARA und Glycerin verantwortlich, wobei drei Serinhydrolasen etwa 99 % der Hydrolyse im Gehirn ausmachen [85]. Etwa 85 % der 2-AG-Hydrolyse erfolgt über die Monoacylglycerol-Lipase (MAGL), die mit CB1-Rezeptoren in Axonterminalen ko-lokalisiert ist [85]. ABHD6 und ABHD12 machen etwa 4 bzw. 9 % der 2-AG-Hydrolase-Aktivität im Gehirn aus, wobei ABHD6 in postsynaptischen Neuronen lokalisiert ist und ABHD12 in Mikroglia hoch exprimiert wird [85]. Die 2-AG-Hydrolyse kann auch durch FAAH katalysiert werden [86]. Die Wege(e) der 2-DPG- und 2-EPG-Hydrolyse sind derzeit unbekannt.
Zusätzlich zu den direkten Signalrollen von 2-AG und AEA sind beide wichtige Zwischenprodukte im Lipidstoffwechsel. Sie dienen als Vorläuferpools für ARA für die nachfolgende Produktion von Eicosanoiden [87] und werden auch in weitere Klassen bioaktiver Mediatoren umgewandelt. 2-AG und AEA sind Substrate für COX-2, die Prostaramide und Prostaglandinglycerolester produziert, LOX, die Hydroperoxyderivate (HPETE) produziert, und CYP-Enzyme, die Hydroxy-Eicosatetraensäure-Ethanolamid-Moleküle (HETE-EA) oder Epoxy-Eicosatriensäuren (EET) produzieren [30, 88, 89]. 2-AG kann auch durch Acylglycerolkinasen zu Lysophosphatidsäure (LPA) phosphoryliert werden [66], einem weiteren wichtigen bioaktiven Lipid [90]. Interessanterweise wurde gezeigt, dass COX-2-Metaboliten von 2-AG und AEA gegensätzliche Effekte zu denen von 2-AG und AEA selbst haben, was auf ein feines Gleichgewicht in der Kontrolle der synaptischen Übertragung zwischen diesen Lipidmediatoren und ihren oxygenierten Produkten hindeutet [91].
Der oxidative Metabolismus von DHA- und EPA-abgeleiteten Endocannabinoiden beginnt geklärt zu werden, aber vieles ist noch unbekannt. Lipidomische Screenings haben oxygenierte Produkte von DHEA identifiziert, die von LOX generiert werden und 10,17-Dihydroxy-Docosahexaenoyl-Ethanolamid (10,17-diHDoHE) und Hydroxy-16(17)-Epoxy-Docosapentaenoyl-Ethanolamid (HEDPEA) umfassen [68]. Diese Moleküle zeigen entzündungshemmende und organschützende Effekte bei einer Maus-Reperfusion-Zweitorganverletzung [68].
Die multiplen Lipidmediatoren, die aus ARA, DHA und EPA stammen, sind in Fig. 2 zusammengefasst, wo zu sehen ist, dass das Lipidom von ARA am besten charakterisiert ist; es werden jedoch wahrscheinlich analoge Repertoires von Mediatoren aus DHA und EPA und potenziell anderen PUFAs produziert. Jüngste Entwicklungen in der lipidomischen Analyse haben das Interesse an der Entdeckung, Identifizierung und Aufklärung der multiplen Mediatoren, die aus PUFA und Endocannabinoiden stammen, stark erhöht, aber es ist noch viel mehr Arbeit erforderlich, um ein vollständiges Verständnis ihrer biologischen Aktivitäten und der Auswirkungen einer Änderung der Nahrungsaufnahme und der nachfolgenden Phospholipid-PUFA-Zusammensetzung auf ihre Bildung zu entwickeln. Der Rest dieser Übersicht wird die aktuelle Evidenz des Zusammenspiels zwischen n-3- und n-6-LCPUFA und Endocannabinoiden bei Neuroinflammation, Neurogenese und Gehirnalterung zusammenfassen.
Fig. 2.
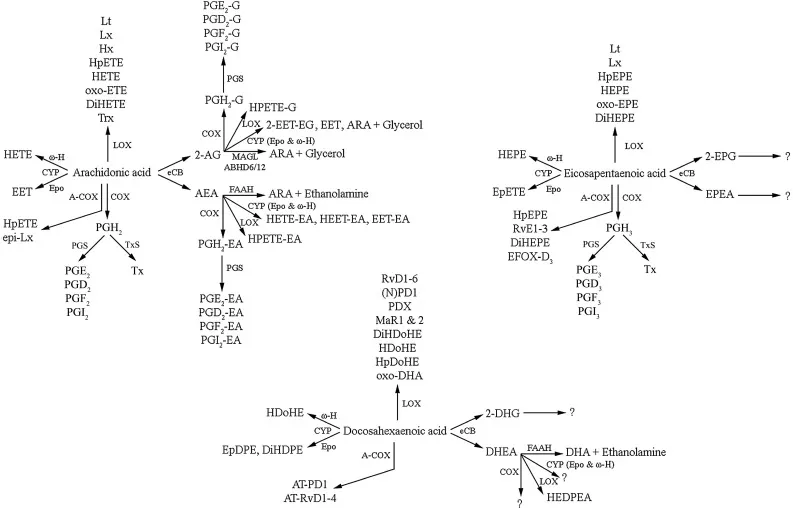 Wichtige Lipidmediatoren aus Arachidonsäure (ARA), Docosahexaensäure (DHA) und Eicosapentaensäure (EPA) im StoffwechselOpen in a new tab
Wichtige Lipidmediatoren aus Arachidonsäure (ARA), Docosahexaensäure (DHA) und Eicosapentaensäure (EPA) im StoffwechselOpen in a new tab
ARA, DHA und EPA sind Vorläufer von multiplen Metaboliten, einschließlich Oxylipinen, die durch Cyclooxygenase (COX) und acetylierte COX-2 (A-COX), Lipoxygenase (LOX) und Cytochrom P450 (CYP) Enzyme sowie die Endocannabinoide (eCB) produziert werden. Die Hauptwege der Synthese von ARA-, DHA- und EPA-abgeleiteten Endocannabinoiden sind in Fig. 3 dargestellt. 2-AG 2-Arachidonoylglycerol, 2-DHG 2-Docosahexaenoylglycerol, 2-EET-EG 2-Epoxy-Eicosatriensäureglycerol, 2-EPG 2-Eicosapentaenoylglycerol, ABHD6/12 α/β-Hydrolase-Domäne 6 oder 12, AEA N-Arachidonoylethanolamin (Anandamid), AT Aspirin-getriggert, DHEA N-Docosahexanoylethanolamin (Synaptamid), DiHDoHE Dihydroxy-Docosahexaensäure, DiHDPE Dihydroxy-Docosapentaensäure, DiHEPE Dihydroxy-Eicosapentaensäure, DiHETE Dihydroxy-Eicosatetraensäure, DiHETrE Dihydroxy-Eicosatriensäure, EDP Epoxy-Docosapentaensäuren, EET Epoxy-Eicosatriensäure, EET-EA Epoxy-Eicosatriensäure-Ethanolamid, EETeTr Epoxy-Eicosatetraensäuren, EFOX Elektrophile Fettsäure-Oxo-Derivate, EPA Eicosapentaensäure, EpDPE Epoxy-Docosapentaensäure, EPEA N-Eicosapentaenoylethanolamin, EpETE Epoxy-Eicosapentaensäure, EpETrE Epoxy-Eicosatriensäure, Epo Epoxygenase, FAAH Fettsäureamidhydrolase, HDoHE Hydroxy-Docosahexaensäure, HEDPEA Hydroxy-Epoxy-Docosapentaenoyl-Ethanolamid, HEET-EA Hydroxyepoxy-Eicosatriensäure-Ethanolamid, HEPE Hydroxy-Eicosapentaensäure, HETE Hydroxy-Eicosatetraensäure, HETE-EA Hydroxy-Eicosatetraensäure-Ethanolamid, HHTrE Hydroxy-Heptadecatriensäure, HpDoHE Hydroperoxy-Docosahexaensäure, HpEPE Hydroperoxy-Eicosapentaensäure, HpETE Hydroperoxy-Eicosatetraensäure, Hx Hepoxilin, Lt Leukotrien, Lx Lipoxin, MAGL Monoacylglycerol-Lipase, MaR Maresin, (N)PD1 (Neuro)protektin D1, NAPE-PLD N-Acyl-Phosphatidylethanolamin-selektive Phospholipase D, NArPE N-Arachidonoyl-Phosphatidylethanolamin, NAT N-Acyltransferase, oxo-EET Oxo-Eicosatetraensäure, PAEA Phospho-Anandamid, PD Protektin, PDE Phosphodiesterase, PE Phosphatidylethanolamin, PGD Prostaglandin-D-Metabolit, PGE Prostaglandin-E-Metabolit, PGF Prostaglandin-F-Metabolit, PGI Prostacyclin, PGS Prostaglandin E, D oder F oder Prostacyclin-Synthase, PI Phosphatidylinositol, PLA1 Phospholipase A1, PLC Phospholipase C, PLD Phospholipase D, PPAR Peroxisom-Proliferator-aktivierter Rezeptor, PTPN22 Protein-Tyrosin-Phosphatase 22, RvD Resolvin-D-Serie, RvE Resolvin-E-Serie, Trx Trioxilin, Tx Thromboxan, TxS Thromboxan-Synthase, SDA Stearidonsäure, SVZ Subventrikulärzone, TRPV-1 Transiente Rezeptorpotenzial-Vanilloidrezeptor Typ 1, ϖ-H ϖ-Hydrolase.
Neuroinflammation
Neuroinflammation ist der ZNS-Prozess zur Wiederherstellung geschädigter Neuronen und Gliazellen, wobei Mikroglia und Astrozyten die vorherrschenden Effektoren sind [92]. Die Aktivierung von Mikroglia leitet eine schnelle Reaktion ein, die Migration, Proliferation und die Freisetzung von Zytokinen und Chemokinen umfasst [93]. Dies ist zunächst eine schützende Reaktion, aber eine überschüssige Neuroinflammation kann die neuronale Regeneration hemmen und, wenn sie chronisch wird, eine wichtige Rolle bei der Pathogenese neurodegenerativer Erkrankungen wie der Alzheimer-Krankheit (AD) und der Parkinson-Krankheit (PD) spielen, indem sie zytotoxische Proteine und reaktive Sauerstoffspezies ausscheidet [94]. Die genaue Rolle von Fettsäuren bei der Entstehung und Linderung von Neuroinflammation wird intensiv erforscht.
Im gesunden Gehirn zeigen Mikroglia einen „Ruhezustand“-Phänotyp, der für die kontinuierliche Immunüberwachung und -beobachtung verantwortlich ist und auch eine Schlüsselrolle bei der Regulierung der neuronalen Plastizität durch Prozesse wie synaptische Pruning und Neurogenese spielt [95]. Pathologische Zustände wie Schäden an neuralen Zellen führen dazu, dass die lokalen „Ruhe“-Mikroglia durch „Aktivierung“ reagieren und schnell ihren Phänotyp ändern und ihre Aktivität umleiten [96]. Je nach Art und Ausmaß der Stimulation wird die Expression spezifischer Gene induziert, die den Mikroglia-Phänotyp entweder auf den klassisch aktivierten (M1) pro-inflammatorischen Phänotyp oder den alternativ aktivierten (M2) anti-inflammatorischen Phänotyp ausrichten [96], obwohl weitere M2a- und M2c-Phänotypen basierend auf dem Induktionsstimulus identifiziert wurden [97].
Arbeiten unseres Labors und anderer haben gezeigt, dass erhöhte n-3-PUFA-Spiegel die Mikroglia-Aktivierung und die anschließende Produktion pro-inflammatorischer Zytokine in einer Vielzahl von Neuroinflammationsmodellen reduzieren, wie z. B. bei amyotropher Lateralsklerose [98], Rückenmarksverletzungen [99, 100], Ischämie [101] und Gehirnalterung [102]. Jüngste Arbeiten haben begonnen, die Mechanismen hinter diesen Effekten zu erforschen. DHA reguliert die Zelloberflächenexpression von Cluster of Differentiation 14 (CD14) und Toll-like Rezeptor 4 (TLR4) in Lipopolysaccharid (LPS)-stimulierten Mikrogliazellen herunter [103]. CD14 ist ein Glycosylphosphatidylinositol-gebundenes Protein und überträgt das Signal durch Assoziation mit anderen Partnern, insbesondere TLR4 [104].
Die n-3-PUFA-Supplementierung hemmt auch die Mikroglia-Aktivierung, indem sie die nukleäre Translokation und Sekretion von High-Mobility Group Box 1 (HMGB1) und die HMGB1-vermittelte Aktivierung von TLR4/NF-κB-Signalwegen in einem Modell für traumatische Hirnverletzungen hemmt [105]. HMGB1 ist ein zentraler Bestandteil der späten Entzündungsreaktion, und die Translokation und Sekretion von HMGB1 sind wichtige Schritte bei der HMGB1-induzierten Entzündung [106]. Nach der Freisetzung bindet HMGB1 an das transmembranäre TLR4 und aktiviert den TLR4/NF-κB-Signalweg, was letztendlich zu Neuroinflammation führt [107]. In dieser Studie hemmte die n-3-PUFA-Supplementierung die Translokation von NF-κB p65 aus dem Zytosol in den Zellkern, reduzierte die NF-κB p65-Expression und hemmte die Expression der mit dem TLR4/NF-κB-Signalweg assoziierten Proteine.
Zusammengenommen deuten diese Ergebnisse darauf hin, dass n-3-PUFAs die Mikroglia-Aktivierung in mehreren Stadien regulieren; diese Effekte könnten jedoch durch die n-PUFA selbst oder ihre jeweiligen SPM vermittelt werden. Beispielsweise blockieren sowohl DHA als auch NPD1 die Zytokinproduktion durch Mikrogliazellen in einer Vielzahl von Netzhaut- und Hirnverletzungsmodellen [108, 109]. RvD1 und MaR1 regulieren die in vitro Mikroglia-Aktivierung herunter [110], RvD2 hemmt den LPS-induzierten Anstieg von TLR4 in Mikroglia [111], und RvE1 verändert die Entzündungsreaktion und reduziert die Mikroglia-Aktivierung in mehreren in vivo-Modellen [112, 113].
Während der Neuroinflammation kommt es zu einer allgemeinen Hochregulierung der Aktivität des Endocannabinoid-Systems, mit überwiegend entzündungshemmenden Effekten [114]. Studien, die die Rolle von Endocannabinoiden bei Neuroinflammation untersuchen, konzentrieren sich jedoch tendenziell auf die Rolle von CB2-Rezeptoren, da CB2-Rezeptoren auf Mikroglia häufiger vorkommen als CB1 [115] und die CB2-Rezeptorexpression in Mikroglia und Astrozyten während der Neuroinflammation erhöht ist [74], wo sie die Freisetzung von Zytokinen aus aktivierten Mikroglia abschwächen [8]. Darüber hinaus zeigen Mikroglia von CB2-Rezeptor-Knockout-Mäusen eine Abnahme der phagozytischen Aktivität, und CB2-Rezeptorantagonisten reduzieren die Motilität von Mikroglia in vitro [116]. Des Weiteren exprimieren Mikroglia in Hirngewebe von Patienten mit Alzheimer-Krankheit (AD), Multipler Sklerose und amyotropher Lateralsklerose CB2-Rezeptoren [115]. Jüngste Arbeiten deuten jedoch auf eine komplexere Situation hin, wobei das Endocannabinoid-System auf den M2-Phänotyp reagiert [117]. CB1- und CB2-Rezeptoren sind in M1-Mikroglia herunterreguliert, während M2a- und M2c-Mikroglia phänotypische Veränderungen in der Endocannabinoid-Maschinerie zeigen, sodass M2a die 2-AG-Synthese und M2c AEA begünstigt. Eine aktuelle Studie hob auch die Rolle von Endocannabinoiden bei der Mikroglia-Neuron-Signalübertragung hervor [118]. Endocannabinoide wurden durch mikrogliale extrazelluläre Membranvesikel ausgeschieden, und diese extrazellulären Vesikel tragen AEA auf ihrer Oberfläche, das CB1-Rezeptoren auf Neuronen stimuliert und die präsynaptische Übertragung hemmt.
Zusätzlich zu Mikroglia reagieren Astrozyten auf ZNS-Schäden und Krankheiten durch den Prozess der „reaktiven Astrogliose“ [119]. In diesem Prozess reagieren Astrozyten auf eine Vielzahl von Zytokinen und Entzündungsmediatoren und produzieren diese auch selbst. Sie interagieren mit einer Reihe von Zelltypen und vermitteln so das Cross-Talk zwischen neuroinflammatorischen und neuralen Systemen [120]. Astrozyten haben auch regulatorische Rollen im PUFA-Stoffwechsel und der Endocannabinoid-Signalübertragung und fördern das Endocannabinoid-Cross-Talk mit anderen Lipidmediatoren. Astrozyten sind in der Lage, ARA und DHA aus LA bzw. ALA zu synthetisieren [121], obwohl die astrocytäre DHA-Synthese viel geringer ist als die DHA-Aufnahme- und Verwertungsraten des Gehirns, was darauf hindeutet, dass die Astrozytensynthese keinen wesentlichen Beitrag leistet [20]. Astrozyten exprimieren MAGL stark, und Mäuse mit spezifischer astrocytärer MAGL-Deletion zeigen mäßig erhöhte 2-AG- und reduzierte ARA-Spiegel sowie reduzierte PGE2- und pro-inflammatorische Zytokinspiegel nach LPS-Verabreichung, was auf eine wichtige Rolle der Astrozyten bei der Endocannabinoid-Signalübertragung bei Neuroinflammation hinweist [122]. Darüber hinaus wurde unter Verwendung eines induzierbaren Knockout-Systems gezeigt, dass der Metabolismus von 2-AG koordiniert von Neuronen und Astrozyten reguliert wird und den transzellulären Transport von Lipidsubstraten wie ARA und Eicosanoiden beinhaltet [123]. Dieser Astrozyten-Neuronen-Cross-Talk könnte eine integrierte Regulation des 2-AG-Metabolismus ermöglichen und eine übermäßige CB1-Rezeptor-Aktivierung verhindern.
Zusammengenommen zeigen diese Studien, dass n-3-PUFA und ihre SPMs sowie 2-AG und AEA wichtige Rollen bei der Regulierung der neuroinflammatorischen Reaktionen von Mikroglia und Astrozyten spielen. Ein besseres Verständnis der Mechanismen, durch die diese Lipidmediatoren miteinander und mit Mikroglia, Astrozyten und umgebenden Neuronen interagieren, könnte jedoch die Entwicklung wirksamer Ansätze zur Regulierung von Neuroinflammation durch Manipulation der diätetischen n-6- und n-3-PUFA-Aufnahme ermöglichen. Um Bauchfett und Entzündungen im Körper zu reduzieren, ist ein ausgewogener Fettsäurenhaushalt von großer Bedeutung.
Lernen, Gedächtnis und synaptische Plastizität
Die Supplementierung mit n-3-PUFA wirkt sich positiv auf viele Aspekte des Lernens und des Gedächtnisses aus, und obwohl eine Reihe mutmaßlicher Ziele identifiziert wurden, sind die genauen Mechanismen, die diesen Effekten zugrunde liegen, noch ungelöst [1]. Eine Studie von Pan und Mitarbeitern legt nahe, dass diese positiven Effekte von der Modulation des Endocannabinoid-Systems abhängen könnten [124]. Das räumliche Gedächtnis von mit DHA behandelten Ratten verbesserte sich bei niedrigeren Dosen (150 oder 300 mg/kg/Tag) signifikant, während es bei einer höheren Aufnahmemenge (600 mg/kg/Tag) beeinträchtigt war. Diese in vivo-dosisabhängigen Effekte korrelierten stark mit ähnlichen in vitro-dosisabhängigen Hochregulationen von CB1- und TRPV-1-Rezeptoren in kultivierten Hippocampus-Neuronen. Die Autoren schlussfolgerten, dass CB1 und TRPV-1 daher an den positiven Effekten der DHA-Supplementierung auf das räumliche Gedächtnis beteiligt sein könnten, obwohl weitere Arbeiten erforderlich sind, um dies zu bestätigen.
Synaptische Plastizität ist ein weit verbreitetes ZNS-Phänomen, das sowohl an erregenden als auch an hemmenden Synapsen auftritt, wo Änderungen in der synaptischen Effizienz und Stärke als Reaktion auf verschiedene Reize induziert werden. Diese Potenzierung oder Depression soll Phänomenen wie Lernen und Gedächtnis zugrunde liegen [125]. Das Endocannabinoid-System moduliert viele Aspekte der synaptischen Plastizität positiv [126], und eine kürzlich durchgeführte elegante Studienreihe von Layė und Mitarbeitern zeigt die essentielle Rolle von n-3-PUFA bei diesen Effekten [13, 14, 127]. In der ersten dieser Studien verhinderte ein langfristiger n-3-PUFA-Mangel die endocannabinoid-vermittelte langfristige synaptische Depression (LTD) im präfrontalen Kortex und im Nucleus accumbens [13]. Cannabinoid-Rezeptoren koppeln an G-Proteine vom Typ Gi/o und aktivieren Signalwege [74], und in dieser Studie waren CB1-Rezeptoren von ihren G(i/o)-Proteinen entkoppelt. In den Folgestudien wurden ähnliche Effekte auf andere Messgrößen der endocannabinoidabhängigen Plastizität auch in anderen Gehirnregionen gefunden, einschließlich des Hypothalamus [14] und des Hippocampus [127]. Im Hippocampus wurde gezeigt, dass der Verlust der N-Methyl-D-Aspartat (NMDA)-Glutamat-Rezeptor-abhängigen LTP, der durch n-3-PUFA-Mangel induziert wurde, auf die Ablation der endocannabinoid-vermittelten inhibitorischen LTD (iLTD) zurückzuführen war [127]. Im Hippocampus wird die LTP durch den Prozess der heterosynaptischen iLTD gesteuert, die von der Aktivierung von CB1-Rezeptoren abhängt [80]. Insgesamt scheint die Rolle der n-PUFA-Regulation des Endocannabinoid-Systems bei Lernen, Gedächtnis und synaptischer Plastizität komplexer zu sein als nur die Modulation der Endocannabinoid-Spiegel, sondern hängt auch entscheidend von der Modulation der Rezeptorfunktion ab.
Neurogenese
Die Neurogenese im erwachsenen Gehirn aus neuronalen Stammzellen wurde konsistent in zwei Regionen identifiziert: der subgranulären Schicht des Hippocampus-Gyrus dentatus und der Subventrikularzone (SVZ). Sie wurde bei allen untersuchten Säugetieren, einschließlich Menschen, berichtet [128]. Der Hippocampus ist unerlässlich für das Lernen und die Gedächtnisbildung und -konsolidierung und spielt auch eine wichtige Rolle bei der Regulierung von Aspekten von Emotionen, Angst, Furcht und Stress [129]. Der Hippocampus ist jedoch besonders anfällig für Neuroinflammation, Alterung und Neurodegeneration [129]; tatsächlich ist das Altern der größte negative Regulator der Hippocampus-Neurogenese [130]. Es ist daher interessant festzustellen, dass die Hippocampus-Neurogenese nach Ischämie [131], Schlaganfall [132] und Anfällen [133] zugenommen hat, wobei die Zunahmen als Versuch des Gehirns zur Selbstheilung betrachtet werden könnten. Die Förderung der Hippocampus-Neurogenese könnte daher einen neuartigen therapeutischen Ansatz bei der Behandlung von Gehirnalterung und Neurodegeneration bieten.
Die Behandlung mit DHA und EPA hat durchweg gezeigt, dass sie die Neurogenese im erwachsenen Hippocampus in einer Reihe von Tiermodellen erhöht [134], auch in neuronalen Stammzellen, wo DHA die neuronale Differenzierung zu fördern scheint [73]. Ähnlich ist das Endocannabinoid-System für die adulte Neurogenese sowohl im Hippocampus [135, 136] als auch in der SVZ [137] unerlässlich, obwohl Studien zu den pro-neurogenen Effekten von Endocannabinoiden im Gyrus dentatus zu widersprüchlichen Ergebnissen geführt haben. Zum Beispiel haben erwachsene Ratten, die mit dem AEA-Analogon Methanandamid behandelt wurden, eine signifikant verminderte Hippocampus-Neurogenese, die durch CB1-Antagonisten erhöht wird [136]. Eine chronische Behandlung mit einem synthetischen Endocannabinoid-Agonisten erhöht jedoch die adulte Hippocampus-Neurogenese bei Ratten [138], und CB1-Rezeptor-Knockout-Mäuse zeigen signifikante Reduktionen der Neurogenese im Gyrus dentatus und der SVZ [135]. Die pharmakologische Blockade von DAGL und CB2 mit spezifischen Antagonisten hemmt die Proliferation neuronaler Stammzellen und die Proliferation von Vorläuferzellen bei jungen Tieren [137]. Eine ähnliche Reaktion wird bei einem FAAH-Inhibitor beobachtet [139]. Insgesamt scheinen die Effekte des Endocannabinoid-Systems auf die Neurogenese ein feines Gleichgewicht der Rezeptoraktivierung zu sein.
Die Arbeit in unserem Labor ist die erste, die die Rolle des Endocannabinoid-Systems bei den pro-neurogenen Effekten von DHA und EPA untersucht [73]. In dieser Studie induziert die Zugabe von DHA oder EPA zu neuronalen Stammzellen gegensätzliche Effekte auf das Zellschicksal, die durch verschiedene Signalwege gesteuert werden. Obwohl sowohl DHA als auch EPA die 2-AG-Spiegel signifikant erhöhen, nutzt nur EPA endocannabinoidale Signalwege, um die Proliferation zu erhöhen. EPA erhöht die Proliferation über CB1/2-Rezeptoren, die den p38-Mitogen-aktivierten Proteinkinase (p38 MAPK)-Signalweg aktivieren. DHA reduzierte die Zellproliferation, was mit der Induktion der Differenzierung übereinstimmt. Es könnte die Hypothese aufgestellt werden, dass, obwohl 2-AG durch DHA erhöht wird, die Effekte durch alternative Wege, wie die Umwandlung in DHEA, gemildert und das Zellschicksal in Richtung Differenzierung gelenkt werden könnten [82]. Rashid und Mitarbeiter zeigen, dass DHEA die Differenzierung neuronaler Stammzellen über Protein-Kinase-A (PKA)/cAMP-Response-Element-Binding-Protein (CREB) induziert. Es könnte daher sein, dass DHA und EPA das Zellschicksal über alternative Wege steuern, die durch die Spiegel und Arten der produzierten Mediatoren bestimmt werden.
Zusätzlich identifizierte unsere Studie eine bisher unerkannte Rolle des Immunsystems bei den Effekten von DHA und EPA [73]. Die DHA- und EPA-Behandlung von neuronalen Stammzellen aus Interleukin-1β (IL-1β)-Knockout-Mäusen induzierte Effekte, die sich deutlich von denen der Wildtyp-Zellen unterschieden, wobei die Proliferation durch DHA erhöht und durch EPA reduziert wurde. Da p38 MAPK durch DHA nicht aktiviert wurde, deutet dies auf alternative nicht-endocannabinoidale Wege hin, die hinter der erhöhten Proliferation steckten.
Das alternde Gehirn
Das normale Gehirnaltern ist durch viele schädliche Veränderungen gekennzeichnet, wie mitochondriale Dysfunktion und Veränderungen im Energiestoffwechsel [140], DNA-Schäden [141], erhöhte Mikroglia-Aktivierung [142] und erhöhter oxidativer Stress [143]. Das alternde Gehirn neigt auch zur Entwicklung neurodegenerativer Erkrankungen wie AD und PD, aber mit den verlängerten prä-symptomatischen Stadien ist es schwierig zu identifizieren, was normale altersbedingte Veränderungen sind und was Effekte einer unentdeckten Neurodegeneration sind [144]. Eine ausgewogene Ernährung, reich an essentiellen Fettsäuren, kann hier präventiv wirken und dazu beitragen, Bauchfett zu verlieren und die allgemeine Gesundheit zu fördern. Auch wenn die Frage, wie man Oberschenkel Fett verlieren kann, oft im Vordergrund steht, ist die Rolle von Fettsäuren für die Gehirnfunktion ebenso wichtig.
Viele epidemiologische Studien deuten auf positive Zusammenhänge zwischen einer erhöhten diätetischen Zufuhr von n-3-PUFA und der Aufrechterhaltung der kognitiven Funktion im Alter hin [3]. Die Ergebnisse randomisierter kontrollierter Studien in diesem Bereich waren jedoch gemischt, obwohl positive Studienergebnisse mit höheren Dosen von DHA, insbesondere bei asymptomatischen Teilnehmern oder solchen mit sehr milden Gedächtnisdefiziten, darauf hindeuten, dass die Supplementierung in der prä-symptomatischen Phase, vor dem Einsetzen einer leichten kognitiven Beeinträchtigung oder Demenz, am wirksamsten ist [145–147].
Studien an Nagetieren und Menschen zeigen, dass das Endocannabinoid-System anfällig für altersbedingte Defizite ist [74]. Zum Beispiel nehmen die CB1-Rezeptorspiegel zusammen mit der Aktivität von NAPE-PLD und DAGL ab [74]. Darüber hinaus führen Abnahmen von DAGL, gekoppelt mit erhöhter MAGL, zu spezifischen Abnahmen der 2-AG-Spiegel im Hippocampus alternder Mäuse [148]. Mithilfe von genetischen CB1-Rezeptor-Knockout-Mausmodellen ist es möglich, die Auswirkungen dieser altersbedingten Veränderungen nachzuahmen [74]. Die CB1-Rezeptor-Deletion führt zu einer altersabhängigen Beschleunigung des kognitiven Verfalls mit beschleunigtem Hippocampus-Neuronenverlust und erhöht Aspekte der Neuroinflammation, wie reaktive Astrogliose und Mikroglia-Aktivierung.
Diese Studien legen nahe, dass der altersbedingte Rückgang spezifischer Komponenten des Endocannabinoid-Systems wichtige Aspekte der Gehirnalterung beschleunigt; daher könnte es durch die Wiederherstellung oder Umkehrung der altersbedingten Effekte möglich sein, diesen Rückgang zu verringern. Neben der Modulation der Spiegel von 2-AG und AEA wurde gezeigt, dass die Expression von CB1-Rezeptoren, TRPV-1 und PPARγ alle auf eine n-3-PUFA-Behandlung ansprechen [79, 124], was darauf hindeutet, dass n-3-PUFA in der Lage sein könnten, einige dieser altersbedingten Verluste zu mildern oder umzukehren. Darüber hinaus könnten diese positiven Effekte auf das Endocannabinoid-System potenziell zu einigen der schützenden Effekte von n-3-PUFA beitragen, die in Studien zum Altern beobachtet wurden. Es ist jedoch noch viel mehr Forschung erforderlich, um unser Verständnis der Mechanismen, die diesen Effekten zugrunde liegen, und der Folgen für das Endocannabinoid-System zu entwickeln, um das therapeutische Potenzial von n-3-PUFA beim Gehirnschutz und der Gehirnreparatur zu maximieren. Im Zusammenhang mit einer gesunden Ernährung ist es auch interessant zu beachten, dass Macadamia gesund sein können und wertvolle Fettsäuren enthalten.
Fazit
Aufgrund ihrer grundlegenden Natur haben ARA, DHA, EPA und ihre Mediatoren sowie das Endocannabinoid-System weitreichende Effekte im gesamten ZNS, und jüngste Erkenntnisse deuten stark auf ein komplexes Zusammenspiel zwischen ihnen hin. Die Spiegel von Phospholipid-gebundener ARA bestimmen die Spiegel von 2-AG und AEA, die zusätzlich zu ihren eigenen biologischen Aktivitäten als ARA-Reservoire für die nachfolgende Eicosanoidproduktion dienen. Wichtig ist, dass die LCPUFA-Spiegel im Gehirn auf die Nahrungsaufnahme reagieren und das n-6:n-3-PUFA-Verhältnis der aktuellen westlichen Ernährung zu einer erhöhten Neuroinflammation und auch einer Überstimulation des Endocannabinoid-Systems führen kann.
Neuroinflammation ist ein Schlüsselmerkmal der Gehirnalterung und Neurodegeneration, und die Entwicklung neuer therapeutischer Ansätze ist notwendig. Epidemiologische Studien zeigen durchweg positive Effekte einer erhöhten Zufuhr von DHA und EPA; diese Beobachtungen haben jedoch bisher nicht zu neuen Behandlungen geführt. Studien liefern typischerweise n-3-PUFA in Form von Fischölen, gemischten DHA- und EPA-Präparaten oder separaten DHA und EPA, mit begrenzter Berücksichtigung der Hintergrundspiegel von n-6-PUFA. Es wird gehofft, dass ein besseres Verständnis der Beziehung zwischen ARA, DHA, EPA und dem Endocannabinoid-System zu Fortschritten bei der Entwicklung ihres therapeutischen Potenzials führen und letztendlich zur Entwicklung gezielterer Behandlungsoptionen für den Gehirnschutz und die Gehirnreparatur beitragen wird.
Abkürzungen
2-AG 2-Arachidonoylglycerol
2-AG-LPA 2-Arachidonoylglycerol-Lysophosphatidsäure
2-AG-LPI 2-Arachidonoyl-Lysophosphatidylinositol
2-DHG 2-Docosahexaenoylglycerol
2-EET-EG 2-Epoxy-Eicosatriensäureglycerol
2-EPG 2-Eicosapentaenoylglycerol
A-COX Acetylierte COX-2
ABHD4 α/β-Hydrolase-Domäne 4
ABHD6 α/β-Hydrolase-Domäne 6
ABHD12 α/β-Hydrolase-Domäne 12
AdA Adrensäure
AEA N-Arachidonoylethanolamin (Anandamid)
ARA Arachidonsäure
AT Aspirin-getriggert
COX-2 Cyclooxygenase-2
CYP Cytochrom P450 Monooxygenase
DAGL Diacylglycerol-Lipase
DGLA Dihomo-γ-Linolensäure
DHA Docosahexaensäure
DHEA N-Docosahexaenoylethanolamin (Synaptamid)
DiHDoHE Dihydroxy-Docosahexaensäure
DiHDPE Dihydroxy-Docosapentaensäure
DiHEPE Dihydroxy-Eicosapentaensäure
DiHETE Dihydroxy-Eicosatetraensäure
DiHETrE Dihydroxy-Eicosatriensäure
DPA Docosapentaensäure
eCB Endocannabinoid
EDP Epoxy-Docosapentaensäure
EET Epoxy-Eicosatriensäure
EET-EA Epoxy-Eicosatriensäure-Ethanolamid
EETeTr Epoxy-Eicosatetraensäuren
EFOX Elektrophile Fettsäure-Oxo-Derivate
EPA Eicosapentaensäure
EpDPE Epoxy-Docosapentaensäure
EPEA N-Eicosapentaenoylethanolamin
EpETE Epoxy-Eicosapentaensäure
EpETrE Epoxy-Eicosatriensäure
Epo Epoxygenase
FAAH Fettsäureamidhydrolase
GP-NAPE Glycerophosphoarachidonoylethanolamid
HDoHE Hydroxy-Docosahexaensäure
HEDPEA Hydroxy-Epoxy-Docosapentaenoylethanolamid
HEET-EA Hydroxy-Epoxy-Eicosatriensäure-Ethanolamid
HEPE Hydroxy-Eicosapentaensäure
HETE Hydroxy-Eicosatetraensäure
HETE-EA Hydroxy-Eicosatetraensäure-Ethanolamid
HHTrE Hydroxy-Heptadecatriensäure
HpDoHE Hydroperoxy-Docosahexaensäure
HpEPE Hydroperoxy-Eicosapentaensäure
HpETE Hydroperoxy-Eicosatetraensäure
Hx Hepoxilin
LCPUFA Langkettige mehrfach ungesättigte Fettsäure
LOX Lipoxygenase
Lt Leukotrien
LTD Langfristige Depression
LTP Langfristige Potenzierung
Lx Lipoxin
MAGL Monoacylglycerol-Lipase
MaR Maresin
(N)PD1 (Neuro)protektin D1
NAPE-PLD N-Acyl-Phosphatidylethanolamin-selektive Phospholipase D
NArPE N-Arachidonoyl-Phosphatidylethanolamin
NAT N-Acyltransferase
oxo-EET Oxo-Eicosatetraensäure
PAEA Phospho-Anandamid
PD Protektin
PDE Phosphodiesterase
PE Phosphatidylethanolamin
PGD Prostaglandin-D-Metabolit
PGE Prostaglandin-E-Metabolit
PGF Prostaglandin-F-Metabolit
PGI Prostacyclin
PGS Prostaglandin E, D oder F oder Prostacyclin-Synthase
PI Phosphatidylinositol
PLA1 Phospholipase A1
PLC Phospholipase C
PLD Phospholipase D
PPAR Peroxisom-Proliferator-aktivierter Rezeptor
PTPN22 Protein-Tyrosin-Phosphatase 22
RvD Resolvin-D-Serie
RvE Resolvin-E-Serie
Trx Trioxilin
Tx Thromboxan
TXS Thromboxan-Synthase
SDA Stearidonsäure
SVZ Subventrikulärzone
TRPV-1 Transiente Rezeptorpotenzial-Vanilloidrezeptor Typ 1
ϖ-H ϖ-Hydrolase
Einhaltung ethischer Standards
Interessenkonflikt
Der Autor erklärt keinen Interessenkonflikt.
Referenzen
- Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function in health and disease. Nat Rev Neurosci. 2014;15(12):771–85.
- Calder PC. Omega-3 fatty acids and inflammatory processes. FASEB J. 2005;19(2):1–19.
- Dangour AD, Whitehouse PJ, Rafferty K, et al. Omega-3 fatty acids for the prevention of dementia. Cochrane Database Syst Rev. 2010;(5):CD005379.
- Gebauer SK, Psota TL, Harris WS, et al. n-3 fatty acid requirement and recommendations for humans. Lipids. 2006;41(1):5–17.
- World Health Organization. Interim summary of conclusions and dietary recommendations on total fat and fatty acids. 2008.
- Food and Agriculture Organization of the United Nations. Fats and fatty acids in human nutrition: report of an expert consultation. 2010.
- Bisogno T, Di Marzo V. Endocannabinoids. Curr Opin Lipidol. 2001;12(5):473–82.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging therapeutic target. Pharmacol Rev. 2006;58(3):389–462.
- Di Marzo V. Endocannabinoids and their signalling in the central nervous system. Nat Rev Neurosci. 2004;5(11):861–72.
- Hillard CJ. The endocannabinoid system and the aging brain. J Gerontol A Biol Sci Med Sci. 2017;72(2):166–73.
- Al Houari Y, Massot-Clavin C, Rault B, et al. Identification of n-3 fatty acid-derived endocannabinoid-like molecules in human plasma. Sci Rep. 2017;7:42981.
- LoVerme J, Laursen WJ, Geci C, et al. Anandamide, anandamide analogs, and DHA-derived anandamide analogs activate the PPARγ receptor. Prostaglandins Leukot Essent Fatty Acids. 2007;76(3):147–53.
- Laye S, Nadjar A, Joffre C, et al. The effect of dietary n-3 fatty acid deficiency on endocannabinoid-mediated neuronal functions in the rat prefrontal cortex. Neuroscience. 2008;156(3):792–801.
- Laye S, Nadjar A, Joffre C, et al. n-3 fatty acid deficiency prevents endocannabinoid-mediated long-term depression in the nucleus accumbens: implications for reward circuitry. Neuropsychopharmacology. 2011;36(1):159–69.
- Laye S, Nadjar A, Joffre C, et al. Differential impact of n-3 fatty acid deficiency on endocannabinoid signaling in the rat hippocampus. Neurobiol Learn Mem. 2014;115:75–84.
- Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood). 2008;233(6):674–88.
- Gibson RA, Neumann MA, Lien EL, et al. Docosahexaenoic acid and arachidonic acid are indispensable for normal brain development. J Nutr. 2013;143(6):790–5.
- Nakamura MT, Nara TY. Essential fatty acid metabolism and its role in disease. Curr Opin Clin Nutr Metab Care. 2004;7(2):135–42.
- Domenici MR, Montosi G, Caporalini G, et al. Liver fatty acid composition and membrane fluidity in rats fed diets supplemented with fish oil. J Nutr Biochem. 2001;12(11):625–32.
- Chang CY, Ke D, Chen JY, et al. Essential fatty acids and the human brain. Acta Neurol Taiwan. 2009;18(4):231–41.
- Cunnane SC, Ryan MA, Bassett K. Brain development and the role of fatty acids. J Pediatr. 2004;145(2 Suppl):S77–81.
- Rapoport SI. Brain arachidonic and docosahexaenoic acid metabolism. Curr Opin Clin Nutr Metab Care. 2008;11(2):137–42.
- Salem N Jr, Litman B, Kim HY, et al. Mechanisms of incorporation of docosahexaenoic acid into brain phospholipids. Lipids. 1999;34(10):1131–8.
- Spector AA, Johansen R, Nielsen MK, et al. Fatty acid transport in the brain. J Lipid Res. 1999;40(6):1069–85.
- Das U, Balaraju T, Reddy PR, et al. Mechanisms of maintaining high arachidonic and docosahexaenoic acid concentrations in brain phospholipids. Prostaglandins Leukot Essent Fatty Acids. 2006;75(4–5):271–8.
- Kjellman P, Östberg F, Lindqvist E, et al. Brain arachidonic acid and docosahexaenoic acid are regulated by distinct mechanisms. Lipids. 2014;49(11):1083–91.
- Niki T, Hino S, Yoshida H. The n-6/n-3 ratio affects brain docosahexaenoic acid (DHA) levels and the expression of genes involved in DHA metabolism in mice. J Nutr Sci Vitaminol (Tokyo). 2013;59(2):161–8.
- Tallima H, El Ridi R. The arachidonic acid cascade: a comprehensive review of its biochemistry, biological roles, and pharmacological modulators. Adv Sci Technol. 2018;2018:7823465.
- Gabbs M, Leng S, Tarimu G, et al. Oxylipins: pro-inflammatory and pro-resolving lipid mediators. Curr Opin Clin Nutr Metab Care. 2015;18(2):179–86.
- Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–22.
- Funk CD, FitzGerald GA. COX-2 inhibitors and the cardiovascular system. Nat Rev Drug Discov. 2007;6(1):1–2.
- Minghetti L. Cyclooxygenase-2 (COX-2) in brain diseases. Curr Drug Targets Inflamm Allergy. 2004;3(4):371–5.
- Nogawa S, Zhang F, Ross ME, et al. Cyclooxygenase-2 expression in the brain after focal ischemia and reperfusion. Brain Res. 1997;756(1–2):259–62.
- Teather LA, Wingerd B, Eisch AJ, et al. Cyclooxygenase-2-mediated neuroinflammation impairs adult hippocampal neurogenesis. Hippocampus. 2006;16(8):666–78.
- Marnett LJ, Rowlinson SW, Goodwin DC, et al. Arachidonic acid binding and hydrolysis by cyclooxygenase-2. J Biol Chem. 1997;272(50):30560–5.
- Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846–52.
- Serhan CN, Chiang N. Novel pro-resolving lipid mediators in inflammation resolution. Curr Opin Pharmacol. 2008;8(3):317–23.
- Mori TA, Beilin LJ, Burke V, et al. Effects of omega-3 fatty acids on blood pressure and heart rate in pregnant women. Hypertension. 2004;43(2):373–9.
- Bagga D, Arbiser J, Howe G, et al. Blockade of cyclooxygenase-2 by celecoxib inhibits angiogenesis and induces apoptosis in human prostate cancer cells. J Clin Oncol. 2004;22(22):4559–68.
- Calder PC, Albers R, Antoine JM, et al. Inflammatory processes and the role of nutrition. Br J Nutr. 2009;101(Suppl 1):S1–S45.
- Laye S, Nadjar A, Joffre C, et al. The effects of dietary n-3 fatty acid deficiency on the brain. Prog Neurobiol. 2011;95(3):395–408.
- Kalupahana NS, Claycombe K, Moustaid-Moussa N. ϖ-3 fatty acids regulate cellular signals of inflammation and insulin sensitivity: a review of mechanisms and clinical implications. Nutr Res. 2011;31(11):773–87.
- Serhan CN, Levy BD. Resolvins in inflammation resolution: signaling mechanisms and therapeutic opportunities. J Clin Invest. 2018;128(2):503–13.
- Bazan NG, Serhan CN. Resolvins, docosatrienes, and neuroprotectins: physiological functions and relevance in neurological disease. J Neurochem. 2004;91(4):755–65.
- Dalli J, Chiang N, Serhan CN. Enhanced resolution of inflammation by novel docosapentaenoic acid-derived resolvins and maresins. FASEB J. 2013;27(7):2574–89.
- Tungen JE, Lund H, Dalli J, et al. Biosynthesis of novel pro-resolving mediators from docosapentaenoic acid (n-3 DPA) in human leukocytes. Front Pharmacol. 2017;8:111.
- Serhan CN. Pro-resolving lipid mediators are functional agonists for the resolution of inflammation. Nature. 2014;510(7503):92–101.
- Lukiw WJ, Cui JG, Marcheselli VL, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in the pathogenesis of Alzheimer’s disease. J Clin Invest. 2005;115(10):2774–83.
- Zhao Y, Lukiw WJ. Neuroprotectin D1: a brain-specific anti-inflammatory and pro-resolving lipid mediator. Front Neurol. 2015;6:146.
- Serhan CN, Chiang N, Dalli J. New pro-resolving mediators in the resolution of inflammation: novel mechanisms for novel therapeutics. J Pathol. 2014;232(2):107–16.
- Schopfer FJ, Cui JG, Cui W, et al. Electrophilic fatty acid oxo-derivatives (EFOX) are a novel class of anti-inflammatory mediators. J Biol Chem. 2011;286(48):41639–49.
- Schopfer FJ, Vita A, Chen W, et al. Electrophilic fatty acid oxo-derivatives (EFOX): a new class of anti-inflammatory lipids. Free Radic Biol Med. 2012;53(9):1644–53.
- Fisslthaler B, Popp R, Kiss L, et al. Nitric oxide activates cGMP-dependent protein kinase to inhibit smooth muscle cell proliferation. J Cell Physiol. 1999;180(1):153–62.
- Spector AA, Johansen R, Nielsen MK, et al. Fatty acid transport in the brain. J Lipid Res. 1999;40(6):1069–85.
- Kunos G, O’Leary DS, Han X, et al. Endocannabinoid system in CNS function and metabolic disorders. Trends Pharmacol Sci. 2009;30(3):141–8.
- Di Marzo V. Endocannabinoid system and the brain: new opportunities for therapeutic intervention. Curr Opin Drug Discov Devel. 2008;11(2):238–46.
- Cristino L, Di Marzo V. The endocannabinoid system in the brain: new avenues for neurological disease therapy. Nat Rev Neurol. 2010;6(1):1–9.
- Al Houari Y, Massot-Clavin C, Rault B, et al. Identification of n-3 fatty acid-derived endocannabinoid-like molecules in human plasma. Sci Rep. 2017;7:42981.
- LoVerme J, Laursen WJ, Geci C, et al. Anandamide, anandamide analogs, and DHA-derived anandamide analogs activate the PPARγ receptor. Prostaglandins Leukot Essent Fatty Acids. 2007;76(3):147–53.
- Kano M, Ohno-Shosaku T, Hashimotodani Y, et al. Endocannabinoid-mediated retro