
Protein-Protein-Interaktionen sind von grundlegender Bedeutung für die Bildung komplexer Interaktionsnetzwerke und den Zusammenbau multimerer Proteinkomplexe, die die funktionalen Arbeitspferde der Zelle darstellen. Ein detailliertes Verständnis der Struktur und Dynamik dieser multimeren und funktionellen Einheiten ist daher entscheidend für das Verständnis ihrer biologischen Funktionen. Die überwiegende Mehrheit der bisherigen Strukturinformationen stammt aus der Röntgenkristallographie und der Kernspinresonanz-Spektroskopie (NMR), die zusammen über 98 % aller Einträge in der Protein Data Bank (PDB) ausmachen (89 % bzw. 9 %). Diese traditionellen Techniken sind jedoch durch ihre Anforderungen an die Probenvorbereitung begrenzt, was ihre Anwendung auf große und dynamische Proteinkomplexe verhindert. Jüngste Fortschritte in der Detektortechnologie der Kryo-Elektronenmikroskopie (Kryo-EM) und bei Algorithmen zur digitalen Bildverarbeitung ermöglichen nun Dichtekarten mit nahezu atomarer Auflösung für die Strukturaufklärung von Proteinkomplexen, doch machen Kryo-EM-Dichtekarten immer noch nur etwa 1 % aller PDB-Einträge aus. Die wachsende Nachfrage nach komplementären Werkzeugen zur Strukturaufklärung hat zur Entwicklung alternativer Methoden zur Charakterisierung von Proteinkomplexen geführt.
Massenspektrometrie (MS)-basierte Strukturtechniken haben im letzten Jahrzehnt ihren Durchbruch erlebt, angetrieben durch die Unfähigkeit traditioneller biophysikalischer Strukturmethoden, die Strukturen und Dynamiken konformationell und zusammensetzungsheterogener Proteinkomplexe aufzulösen. Methoden wie kovalente Markierung/Footprinting [1, 2], Wasserstoff-Deuterium-Austausch [3], Vernetzungs-Massenspektrometrie (XL-MS, manchmal abgekürzt CX-MS, CL-MS) [4–7], Ionenmobilitäts-MS [8] und native MS [9] sind heute wertvolle Bestandteile der Werkzeugkiste des Strukturbiologen und ermöglichen die Charakterisierung von Proteinkomplexen mit geringer Auflösung, die der Charakterisierung mit traditionellen Mitteln entgangen sind. Obwohl sie einzeln nicht in der Lage sind, eine vollständige Struktur zu liefern, liegen die Vorteile dieser Ansätze in ihrer Vielseitigkeit, Empfindlichkeit und ihrem Durchsatz, die es ihnen ermöglichen, nützliche Informationen zur Ergänzung konventioneller Strukturanalysen bereitzustellen. In Kombination mit Strukturwerkzeugen wie Röntgenkristallographie und Elektronenmikroskopie ermöglichen Hybridtechniken die architektonische Aufklärung multimerer Proteinkomplexe, die sich allein traditionellen Methoden widersetzt haben. Diese MS-basierten Strategien und ihre Beiträge zur Strukturbiologie wurden in den letzten Jahren ausführlich rezensiert [10–28]. Unter diesen Werkzeugen ist die XL-MS unter den MS-basierten Techniken einzigartig, da sie die Fähigkeit besitzt, Protein-Protein-Interaktionen (PPI) aus ihrer nativen Umgebung gleichzeitig zu erfassen und ihre physikalischen Interaktionskontakte aufzudecken, wodurch die Bestimmung sowohl der Identität als auch der Konnektivität von Protein-Protein-Interaktionen in Zellen ermöglicht wird [29, 30].
Der allgemeine XL-MS-Workflow ist in Abbildung 1 dargestellt. Kurz gesagt, Proteine werden zunächst mit bifunktionellen Vernetzungsreagenzien umgesetzt, die räumlich nahe beieinander liegende Aminosäurereste durch die Bildung kovalenter Bindungen physikalisch verknüpfen. Die vernetzten Proteine werden dann enzymatisch verdaut, und die resultierenden Peptidgemische werden getrennt und mittels Flüssigkeitschromatographie-Tandem-Massenspektrometrie (LC-MS/MS) analysiert. Die anschließende Datenbanksuche von MS-Daten identifiziert vernetzte Peptide und ihre Verknüpfungsstellen. Da jedes Vernetzungsreagenz eine definierte Länge besitzt, können die resultierenden Vernetzungen als Abstandsrestriktionen für verschiedene Anwendungen genutzt werden, die von der Strukturvalidierung und integrativen Modellierung [31–36] bis zur de novo-Strukturvorhersage [35, 37, 38] reichen. In den letzten Jahren haben signifikante technologische Fortschritte in XL-MS-Studien das Feld dramatisch vorangetrieben und eine breite Palette von Anwendungen in vitro und in vivo ermöglicht, nicht nur auf der Ebene von Proteinkomplexen, sondern auch im Proteommaßstab. Mehrere aktuelle Übersichtsartikel haben sich auf spezifische Aspekte der XL-MS konzentriert, darunter Reagenzdesign [39] und Anwendungen [12, 19, 27, 28, 40–42]. Diese Übersicht bietet einen Überblick über die jüngsten Entwicklungen in XL-MS-Studien, insbesondere in den letzten drei Jahren. Insbesondere beschreiben wir Verbesserungen bei Vernetzungsreagenzien, Probenvorbereitung, XL-MS-Workflows und Bioinformatik-Tools, um nicht nur die inhärenten Herausforderungen der XL-MS anzugehen, sondern auch das Spektrum der Vernetzungsstudien zu erweitern. Darüber hinaus werden repräsentative in vitro– und in vivo-Anwendungen beschrieben, um die Wirksamkeit und das Potenzial der XL-MS bei der Definition von Proteininteraktionslandschaften und Architekturen großer Proteinkomplexe zu veranschaulichen. Ferner wird der aktuelle Status der komparativen XL-MS skizziert, um deren Rolle bei der Untersuchung von Proteininteraktionen und Strukturdynamiken zu exemplifizieren. Schließlich werden Zukunftsperspektiven für XL-MS-Strategien der nächsten Generation erörtert. Aufgrund des begrenzten Platzes in dieser Übersicht bitten wir um Entschuldigung, dass nicht die gesamte in den letzten Jahren veröffentlichte Literatur berücksichtigt werden konnte.
1. Strategien zur Überwindung inhärenter Herausforderungen in XL-MS-Studien
Obwohl die XL-MS-Analyse mehrere Jahrzehnte zurückreicht [43], hat sich diese Technik in den letzten Jahren endlich zu einem zugänglichen und leistungsstarken Strukturwerkzeug zur Kartierung von Protein-Protein-Interaktionen entwickelt. Ihre Wirksamkeit wurde lange Zeit durch drei primäre Hindernisse beeinträchtigt: 1) komplexe MS/MS-Fragmentierung von vernetzten Peptiden; 2) geringe Häufigkeit von vernetzten Peptiden in komplexen Peptidgemischen; 3) Heterogenität der vernetzten Produkte. Während die erste Hürde die genaue Identifizierung vernetzter Peptide und die eindeutige Zuordnung vernetzter Stellen erschwert, behindern die letzteren die effektive MS-Detektion vernetzter Peptide. Im Laufe der Jahre wurden enorme Anstrengungen unternommen, um diese Herausforderungen zu überwinden, was zu verschiedenen technologischen Fortschritten bei XL-MS-Analysen führte, wie unten hervorgehoben wird. Diese Innovationen haben die Realisierung des Potenzials der XL-MS in der heutigen Strukturbiologie kollektiv gefördert.
1.1 Bioinformatik-Tools zur Analyse vernetzter Peptide
1.1.1 Identifizierung vernetzter Peptide
Die MS/MS-Fragmentierung konventioneller vernetzter Peptide ist typischerweise komplex und unvorhersehbar, was spezialisierte Algorithmen oder Software erfordert, um die Identitäten beider Einzelpeptide innerhalb einer Vernetzung genau zu sequenzieren. Aus bioinformatischer Sicht stellt dies auch ein Skalierungsproblem dar, da die meisten Datenbank-Suchplattformen funktionieren, indem sie experimentelle MS/MS-Spektren mit einer berechneten Bibliothek theoretischer Spektren vergleichen. Bei vernetzten Peptiden nimmt der Suchraum exponentiell statt linear zu, da alle möglichen Peptidkombinationspaare (n2) beim Aufbau einer Bibliothek berücksichtigt werden müssen [44, 45]. Dieses Problem erhöht schnell die Rechenleistung und -zeit, die für die Identifizierung vernetzter Peptide erforderlich sind, wenn die Gesamtzahl der Proteine in einer Probe zunimmt.
Um diese Probleme zu adressieren, wurden verschiedene Softwarepakete entwickelt, die die Datenbanksuche zur Identifizierung vernetzter Peptide ermöglichen, wie in Tabelle 1 zusammengefasst. Eine gängige Strategie besteht darin, spezielle Algorithmen zur Reduzierung des Suchraums zu verwenden, um die Rechenlast zu verringern. Zum Beispiel wurde xQuest/xProphet entwickelt, um groß angelegte Datenbankrecherchen zu ermöglichen, indem eine isotopenbasierte Kandidatenpeptid-Vorfilterung verwendet wird, um die Anzahl der zu berücksichtigenden Permutationen zu minimieren [46, 47]. StavroX vergleicht Präkursorionenmassen mit einer vorab berechneten Liste theoretischer Vernetzungen und verwendet die Masse, um potenzielle Übereinstimmungen zu korrelieren [48]. Algorithmen wie das schnelle Sortieren [45] basieren ebenfalls auf der Massenfilterung, nutzen jedoch die Array-Sortierung, um die Anzahl der erforderlichen Iterationen während des Vorgangs zu reduzieren, was zu schnelleren Suchen führt. Im Vergleich dazu behandeln Software wie ProteinProspector [49, 50] und pLink [44] vernetzte Peptide als einzelne Peptide mit großen, unbekannten Modifikationen, identifizieren einzelne Peptid-Treffer und rekombinieren dann diejenigen, die aus gemeinsamen Spektren stammen, zu vernetzten Treffern.
In den letzten Jahren wurden verschiedene Software-Suiten entwickelt, um die Cross-Link-Identifizierung zu verbessern. Suchmaschinen wie SIM-XL [51] und Kojak [52] bauen auf Strategien zur Minimierung des Suchraums durch heuristische Ansätze auf. SIM-XL kann Möglichkeiten eliminieren, indem nur Cross-Link-Kombinationen berücksichtigt werden, die mindestens ein Peptid enthalten, das mit einer Dead-End-Modifikation identifiziert wurde, und indem nur Spektren durchsucht werden, die charakteristische Ionen aufweisen, die ausschließlich von Cross-Linked-Peptiden stammen (wie solche, die aus Lysin-Seitenketten-Umlagerungen nach der Vernetzung resultieren) [53]. Kojak wendet einen Zwei-Pass-Algorithmus an, der zunächst nach einzelnen Kandidatenpeptiden mit Modifikationen sucht, die der Differenz zwischen Präkursorion und Peptidmassen entsprechen, und dann die besten Peptide aus jedem Spektrum paart, um nach Cross-Linked-Peptiden basierend auf der Präkursorionmasse zu suchen [52]. Im Vergleich dazu wurden andere Tools entwickelt, um alle Kombinationen von Peptidpaaren zu durchsuchen. Xilmass generiert eine Bibliothek von Spektren für alle potenziellen Cross-Links und deren Fragmentionen, was seine Verwendung auf einzelne Komplexe beschränkt, da die für komplexere Suchen erforderliche Zeit zu hoch wäre [54]. XLSearch verwendet eine probabilistische Bewertungsmethode und maschinelles Lernen, um die Genauigkeit der Cross-Link-Identifizierung zu verbessern [55]. Schließlich verwenden ECL/ECL2 einen neuartigen Algorithmus, der alle Peptidkombinationen mit linearer Zeit- und Raumkomplexität erschöpfend durchsucht, was eine ungefilterte Analyse von Cross-Linked-Peptiden mit großen Datenbanken innerhalb weniger Stunden ermöglicht [56, 57]. Wie gezeigt, verwenden jede Software einzigartige Kombinationen von Algorithmen und Regeln, um die besten Cross-Link-Identifikationen zu liefern (Tabelle 1).
1.1.2 Automatisierte Visualisierung vernetzter Reste
Jüngste Bioinformatik-Tools wurden auch entwickelt, um die automatisierte Visualisierung von Vernetzungsdaten durch die Generierung von zweidimensionalen vernetzten Restnetzwerken oder die Abbildung von Vernetzungen auf dreidimensionale Strukturen zu ermöglichen. xVis [58], xiNET [59], ProXL [60] und CLMSVault [61] sind Beispiele für kürzlich entwickelte Software, die die Visualisierung von Vernetzungsdaten als Netzwerke verbundener Reste ermöglichen. Darüber hinaus erleichtert CLMSVault [61] die Abbildung von Vernetzungen im Kontext dreidimensionaler Strukturen, ähnlich wie ProXL [60] und XlinkDB 2.0 [62], die auch als öffentliche Repositorien für Vernetzungsdaten fungieren. Während die meisten Mapping-Tools euklidische Distanzen zur Bestimmung der Vernetzungslänge verwenden, wurden Xwalk und Jwalk entwickelt, um lösungsmittelzugängliche Oberflächendistanzen (SASD) für Vernetzungen zu bestimmen, d.h. die Länge des kürzesten Pfades zwischen zwei Aminosäuren in Bezug auf das Proteinvolumen [63, 64]. Tools wie XLMap können verwendet werden, um Proteinmodelle basierend auf der Vernetzungszufriedenheit zu bewerten und zu evaluieren [65]. Zusammen ermöglichen diese automatisierten Tools (Tabelle 1) die Visualisierung von Vernetzungsdaten innerhalb primärer Sequenzen und bestehender Strukturen, wodurch die Dateninterpretation bei der Strukturaufklärung erleichtert wird. Für die Verwaltung und Analyse großer Datensätze kann der Einsatz von leistungsfähigen Tabellenkalkulationsprogrammen und excel fortbildung online von Vorteil sein, um auch komplexe biologische Informationen effizient zu verarbeiten.
1.2 Entwicklung MS-spaltbarer Vernetzungsreagenzien
Neben der Softwareentwicklung haben große Anstrengungen zur Erleichterung der Identifizierung und Charakterisierung vernetzter Peptide in der Entwicklung neuer Vernetzungsreagenzien, nämlich spaltbarer Vernetzer [39], gegipfelt. Diese Reagenzien tragen labile Bindungen, die aufgrund ihrer einzigartigen chemischen Eigenschaften auf verschiedene Weisen gespalten werden können, z. B. durch Photo- [66], chemisch- [5, 67] und MS-induzierte Spaltungen [68–80]. Da die spaltbaren Bindungen innerhalb der Spacerregionen der Linker lokalisiert sind, können die beiden vernetzten Peptidbestandteile entweder vor oder während der MS-Analyse physikalisch getrennt werden. Während chemisch-induzierte und photolabile Vernetzungsreagenzien die Cross-Link-Trennung vor der MS-Analyse ermöglichen [66, 81–83], fragmentieren MS-spaltbare Reagenzien innerhalb des Massenspektrometers, wodurch eine effiziente Korrelation zwischen getrennten Cross-Link-Peptidbestandteilen und ihren jeweiligen Mutterionen ermöglicht wird. Dieses einzigartige Merkmal von MS-spaltbaren Reagenzien ermöglicht die Produktion charakteristischer Cross-Link-Fragmente in MS2, wodurch die nachfolgende MS-Analyse und Datenverarbeitung zur einfachen und genauen Identifizierung vernetzter Peptide vereinfacht wird. Diese kombinierten Vorteile machen MS-spaltbare Vernetzungsreagenzien zum attraktivsten Typ spaltbarer Reagenzien für XL-MS-Studien.
Mehrere Klassen spaltbarer Bindungen wurden in aktuelle MS-spaltbare Vernetzer integriert, wie in Tabelle 2 zusammengefasst. Unter ihnen sind CID-spaltbare Bindungen am beliebtesten, da sie in der Mehrheit der bestehenden MS-spaltbaren Reagenzien vorhanden sind. Wie gezeigt, sind die drei häufig verwendeten CID-spaltbaren Bindungen: 1) C-S-Bindungen neben Sulfoxid [74, 79, 84, 85]; Sulfonium-Ion [70] oder Cyanurring [75]; 2) D-P (Asp-Pro)-Bindungen [86–88]; 3) C-N-Bindungen, die mit Rink- [68, 76, 89], Harnstoff- [73, 90, 91] oder quaternären Diaminostrukturen [92] assoziiert sind. Abhängig von der Stärke der spaltbaren Bindungen können diese Reagenzien für MSn- und/oder MS2-basierte Workflows implementiert werden, um die Identifizierung vernetzter Peptide zu erleichtern, wie in Tabelle 2 detailliert beschrieben.
Tabelle 2. Repräsentative MS-spaltbare Vernetzer
| Spaltbare Bindung | Dissoziationstyp | Reagenz | Workflow # | Refs |
|---|---|---|---|---|
| C-S (Sulfoxid) | CID | MSn (CID2, CID3)MS2 (CID2, ETD2)MS2 + MSn | [74, 95][101][102] | |
| MSn (CID2, CID3) | [84,96] | |||
| MSn (CID2, CID3) | [85] | |||
| MSn (CID2, CID3) | [79, 93] | |||
| C-S (Sulfonium) | CID | MSn (CID2, CID3) | [70] | |
| C-S (Cyanursäure) | CID | MS2 (CID2) | [75] | |
| D-P (Asp-Pro) | CID | MS2 (IS1, CID2)MSn (CID2, CID3) | [69, 86] | |
| MSn (CID2, CID3) | [89, 103] | |||
| CID/ETD | MSn (CID2, CID3)ETD2 Validierung | [88] | ||
| C-N (Rink) | CID | MSn (CID2, CID3) | [68, 87] | |
| MSn (CID2, CID3) | [76] | |||
| C-N (Harnstoff) | CID | MS2 (CID) | [73] | |
| MS2 (HCD) | [90] | |||
| MS2 (HCD) | [91] | |||
| C-N (Quaternäres Diamin) | CID | MSn (CID2, CID3) | [92] | |
| N=N | CID (FRIPS) | MSn (HCD2, CID3) | [112] | |
| N-N (Hydrazon) | ETD | MSn (ETD2, CID3) | [71] | |
| C-N | ETD | MS2 (ETD2) | [72] |
1.2.1 MSn-basierter XL-MS-Analyse-Workflow
Die idealen Vernetzer für die MSn-Analyse sollten MS-spaltbare Bindungen besitzen, die signifikant labiler sind als Peptidbindungen, um eine selektive und bevorzugte Fragmentierung des Linkers mit minimaler Peptid-Rückgratspaltung im MS2-Stadium zu gewährleisten. Eine solche Fragmentierung sollte auch unabhängig von Peptidladung und -sequenz erfolgen. Zu diesem Zweck haben wir eine Reihe von sulfoxidhaltigen, MS-spaltbaren Vernetzern (d.h. DSSO [74], DMDSSO [84], Azide/Alkyne-A-DSBSO [79, 93] und DHSO [85]) entwickelt und gezeigt, dass die C-S-Bindungen neben dem Sulfoxid robuste MS-labile Bindungen sind, die die gewünschten Merkmale für die MSn-Analyse aufweisen (Tabelle 2). Es ist zu beachten, dass die CID-Spaltbarkeit der C-S-Bindungen in einem sulfoniumionenhaltigen und CBDPS-Vernetzer ebenfalls illustriert wurde [70, 75]. Während unsere sulfoxidhaltigen Vernetzer [74, 79, 84, 85, 93] jeweils unterschiedliche chemische Merkmale aufweisen, die für spezifische Anwendungen konzipiert wurden, sind sie alle homobifunktionelle Vernetzer mit zwei symmetrischen CID-spaltbaren C-S-Bindungen. Infolgedessen erfolgt die Identifizierung aller sulfoxidhaltigen MS-spaltbaren vernetzten Peptide unter Verwendung derselben MSn-basierten Analyseplattform, wie in Abbildung 2A dargestellt.
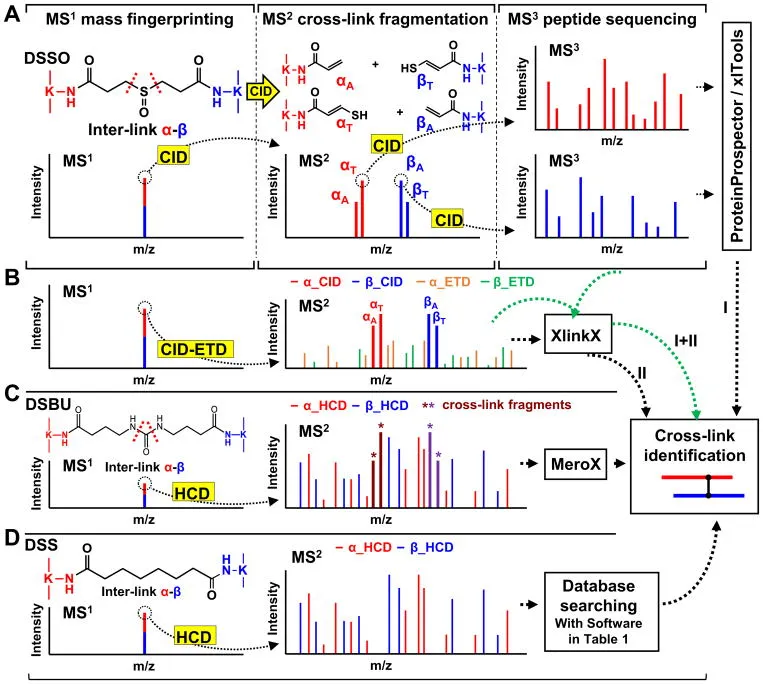 Workflows für die MS-Analyse von MS-spaltbaren (A–C) und nicht spaltbaren vernetzten Peptiden (D).
Workflows für die MS-Analyse von MS-spaltbaren (A–C) und nicht spaltbaren vernetzten Peptiden (D).
Unter Verwendung eines DSSO-verknüpften Inter-Peptids α-β als Beispiel (Abbildung 2A) induziert die niederenergetische CID während MS2 die Spaltung einer der beiden symmetrischen C–S-Bindungen neben der Sulfoxid-Funktionsgruppe, was zur physikalischen Trennung der Vernetzung führt und einzigartige Peptidfragmentpaare (d.h. αA/βS oder αS/βA) mit einer definierten Massenbeziehung liefert [74]. Die resultierenden α- und β-Peptidfragmente sind mit komplementären Vernetzer-Restmolekülen modifiziert, d.h. Alken (A) oder Sulfensäure (S). Die Sulfensäure-Einheit unterliegt jedoch oft einer Dehydratisierung, um eine stabilere ungesättigte Thiol-Einheit (T) zu bilden. Diese charakteristischen und vorhersagbaren MS2-Fragmentionenpaare (d.h. αA/βT und αT/βA) werden dann einer MS3-Analyse unterzogen, um vernetzte Peptide durch konventionelle Datenbank-Suchwerkzeuge vereinfacht und eindeutig zu identifizieren. Dieser Workflow ermöglicht die Datenbanksuche im Proteom-Maßstab mit dem gleichen Maß an Geschwindigkeit, Vertrauen und Genauigkeit, das in routinemäßigen Proteomikstudien geboten wird. Die Falsch-Erkennungsrate (FDR) von Vernetzungen wird durch die Strenge, die mit der Einbeziehung von drei Beweislinien (d.h. MS1-Massenkartierung, MS2-Vernetzungsfragmentierung, MS3-Peptidsequenzierung) verbunden ist, weiter verringert. Zusätzlich zur konventionellen Top-N-Akquisition während der MS3-Analyse kann eine gezielte Akquisition effektiv implementiert werden, indem die Massendifferenz von Alken- und Thiol-modifizierten Fragmentpeptiden genutzt wird (z.B. für DSSO-vernetzte Peptide: Δm = (αT-αA) = (βT-βA) = 31,97 Da) [74, 94]. Beide Arten von Akquisitionsmethoden liefern vergleichbare und komplementäre Ergebnisse bei datenabhängigen Analysen. Dieser MSn-basierte Workflow wurde erfolgreich angewendet, um Protein-Protein-Interaktionen zu kartieren und Architekturen von Proteinkomplexen in vitro und in vivo aufzuklären [74, 79, 95–100].
Obwohl effektiv, leidet die MSn-Analyse typischerweise unter geringerer Empfindlichkeit und erfordert längere Zykluszeiten im Vergleich zu MS2-basierten Akquisitionen. Die Empfindlichkeit und Scangeschwindigkeit der MSn-Analyse wurden jedoch durch die Entwicklung neuer Generationen von Orbitrap-Massenspektrometern erheblich verbessert. Es ist klar, dass fortgeschrittene MS-Instrumente dazu beitragen, XL-MS-Studien voranzutreiben, indem sie die MSn-basierte Identifizierung von gering abundanten vernetzten Peptiden aus zunehmend komplexen biologischen Proben verbessern [79, 98, 99]. Mit der Verfügbarkeit neuer Fragmentierungstechniken in neuen Instrumenten hat die Heck-Gruppe kürzlich einen MS2-basierten Workflow unter Verwendung sequenzieller CID-ETD [101, 102] zur Sequenzierung von DSSO-vernetzten Peptiden demonstriert (Abbildung 2B). Da während der CID in MS2 nur eine begrenzte Rückgratfragmentierung von DSSO-vernetzten Peptiden auftritt, wird ETD durchgeführt, um die Peptidfragmentierung zu verstärken und bei der Sequenzidentifizierung zu helfen [74]. Die Software XlinkX wurde speziell für diesen Zweck entwickelt und nutzt alle Formen von Fragmentinformationen in MS2 für die Datenbanksuche (Tabelle 1) [101, 102]. Ähnlich dem MSn-Workflow ist das Vorhandensein vorhersagbarer und dominanter DSSO-Cross-Link-Fragmente essentiell für die genaue Identifizierung vernetzter Peptide aus MS2-Daten. Während DSSO-basierte MSn- [95, 98, 99] und MS2-Workflows [101] jeweils ihre Wirksamkeit bei der Identifizierung vernetzter Peptide aus verschiedenen komplexen Proben demonstriert haben, hat sich die Integration von MSn- und MS2-Strategien als vielversprechender erwiesen, um umfassendere und komplementäre Informationen für systemweite Studien zu liefern [102].
Eine weitere repräsentative Gruppe von MSn-basierten MS-spaltbaren Reagenzien sind die PIR (Protein Interaction Reporter)-Vernetzer, die vom Bruce-Labor entwickelt wurden und duale Rink- [68] oder D-P-Bindungen [89] für die CID-induzierte Spaltung nutzen. Zusätzlich zur charakteristischen Cross-Link-Fragmentierung wird ein Reporterion gebildet und in MS2 detektiert. Um die Auswahl nur vernetzter Peptidfragmente für die nachfolgende MS3-Sequenzierung sicherzustellen, wurde ReACT (Real-time Analysis for Cross-linked peptide Technology) auf der Grundlage einzigartiger Merkmale von PIR-Vernetzer entwickelt, um eine Echtzeit-Entscheidungsfindung zu ermöglichen [77, 103]. Gekoppelt mit Biotin-basierter Anreicherung wurden PIR-Reagenzien erfolgreich für verschiedene groß angelegte Vernetzungsanwendungen in Bakterien [77, 89, 104–106], Murinen [107] und menschlichen Systemen [40, 77, 108] eingesetzt. Kollektiv sind MSn-basierte Workflows unter Verwendung von CID-spaltbaren Reagenzien effektiv und robust zur Kartierung von PPIs aus verschiedenen Arten komplexer Proben.
1.2.2 MS2-basierter XL-MS-Workflow
Für die MS2-basierte Analyse wären die Voraussetzungen für MS-spaltbare Bindungen weniger streng als die für die MSn-Analyse entwickelten. Eine charakteristische Fragmentierung der Vernetzungen ist jedoch weiterhin erforderlich, da die diagnostischen Ionen, die aus der Spaltung der Vernetzung entstehen, eine genauere Identifizierung vernetzter Peptide auf MS2-Ebene gewährleisten als die Identifizierung nicht spaltbarer vernetzter Peptide (Abbildung 2C und D). Im Vergleich zu MSn sind MS2-basierte Workflows empfindlicher und weisen eine höhere Scangeschwindigkeit auf, wodurch die Wahrscheinlichkeit der Sequenzierung vernetzter Peptide erhöht wird. Einige inhärente Probleme, die mit der MS2-basierten Vernetzungsanalyse verbunden sind, bleiben jedoch bestehen; Fragmentionen sind für zwei Peptidsequenzen vorhanden, und eine ungleiche Fragmentierung kann zu unzureichenden Informationen führen, um beide Peptide gleichzeitig genau zu identifizieren. Daher wäre die Datenbanksuche von MS2-Daten – selbst für MS-spaltbare Vernetzer – geschwindigkeitsbegrenzend und anfälliger für höhere FDR im Vergleich zur MSn-basierten Analyse. Um falsch positive Ergebnisse zu eliminieren, ist Vorsicht geboten und eine strenge Filterung muss implementiert werden [109]. Nichtsdestotrotz ist die MS2-basierte Analyse aufgrund ihrer Empfindlichkeit und Geschwindigkeit sowie ihrer Instrumentenflexibilität und Zugänglichkeit attraktiv. Um die Datenanalyse und -verwaltung in diesem Kontext zu optimieren, sind solide Kenntnisse in der Datenverarbeitung und -organisation unerlässlich, was durch eine gezielte excel fortbildung erreicht werden kann.
Wie in Tabelle 2 aufgeführt, eignen sich mehrere MS-spaltbare Vernetzer besser für die MS2-Analyse, da ihre MS-spaltbaren Bindungen vergleichbare Stärken wie Peptidam-Bindungen aufweisen. Ihre Spaltung erfordert daher eine Kollisionsenergie, die der für die Fragmentierung von Peptidrückgraten benötigten ähnelt. Ein solcher bemerkenswerter Vernetzer ist der Harnstoff-basierte MS-spaltbare homobifunktionale NHS-Ester, d.h. DSBU (auch bekannt als BuUrBu), der von der Sinz-Gruppe entwickelt wurde und spaltbare C-N-Bindungen neben einer zentralen Harnstoff-Funktionsgruppe enthält [73]. Die charakteristische Fragmentierung von DSBU (Abbildung 2C) unterstützt die Datenbanksuche von MS2-Spektren durch die MeroX-Software (Tabelle 1) [110, 111]. Um die genaue Zuordnung vernetzter Produkte durch MS2-Analysen weiter zu erleichtern, wurde das DSBU-Derivat Bromphenylharnstoff (BrPU) entwickelt, indem zwei Bromatome in die Linkerregion integriert wurden. Diese Integration ermöglicht die Detektion des einzigartigen Brom-Isotopenmusters und Massendefekts für alle mit Vernetzern modifizierten Fragmente, was die automatisierte Analyse und Zuordnung vernetzter Peptide durch MeroX verbessert [90].
Ganz kürzlich hat die Sinz-Gruppe entdeckt, dass CDI (1,1′-Carbonyldiimidazol), eine kommerziell erhältliche Verbindung, zwei symmetrische harnstoffartige MS-spaltbare Bindungen mit einer ultrakurzen Spacerlänge (~2,6 Å) trägt (Tabelle 2) und somit als „Null-Längen“-MS-spaltbarer Vernetzer fungieren kann [91]. Es wurde gezeigt, dass CDI Amin- und Hydroxylgruppen mit ähnlicher Reaktivität nahe dem physiologischen pH-Wert angreift, was die Abdeckung von Proteininteraktionen erweitert. Darüber hinaus fragmentieren CDI-vernetzte Peptide während der CID in MS2 ähnlich wie andere harnstoffhaltige MS-spaltbare Vernetzer, wobei charakteristische Produktionen zur Erleichterung der Vernetzungsidentifizierung entstehen [91]. Zudem wird die Heterogenität der vernetzten Produkte durch das Fehlen von „Dead-End“-modifizierten Peptiden (Typ 0) reduziert.
1.2.3 Andere MS-spaltbare Vernetzer
Zusätzlich zu den oben beschriebenen normalen CID-spaltbaren Bindungen wurden kürzlich FRIPS (Free Radical Initiated Peptide Sequencing)-basierte Vernetzer untersucht [78, 80, 112]. Im Vergleich zu TEMPO- [80] und Azo-Linkern [78] scheint der ABI (Azobisimidoester)-Linker (Tabelle 2) am vielversprechendsten zu sein, da er den FRIPS-Prozess während der HCD im Positivionenmodus induzieren kann [112]. Obwohl interessant, scheint ihre Leistung für die automatisierte Identifizierung vernetzter Peptide aufgrund komplexer Fragmentierung weniger günstig zu sein als die normalen CID-spaltbaren Vernetzer [90]. Während die meisten MS-spaltbaren Vernetzer CID-spaltbar sind, wurden auch ETD-spaltbare Bindungen für Vernetzungsstudien verwendet (Tabelle 2) [71, 72]. Basierend auf bestehenden spaltbaren Bindungen wurde der dual-spaltbare Vernetzer DUCCT entwickelt, der sowohl CID-spaltbare als auch ETD-spaltbare Stellen enthält [88]. Die Integration von zwei differentiellen spaltbaren Bindungen ermöglicht komplementäre Vernetzungsfragmentierungen, die durch separate CID- und ETD-Analysen in MS2 erhalten werden, um die Vernetzungsidentifizierung zu unterstützen. Obwohl eine effektive ETD-Analyse im Allgemeinen hochgeladene Präkursorionen erfordert, eignen sich vernetzte Peptide für die ETD-Analyse, da sie aufgrund des Vorhandenseins von zwei tryptischen Peptiden mindestens 4+ Ladungen tragen. In der Vergangenheit hatte die ETD-Analyse jedoch im Vergleich zur CID eine signifikant geringere Empfindlichkeit. Die Implementierung von Hochleistungs-ETD im Orbitrap Fusion Lumos MS hat eine stark verbesserte Empfindlichkeit gezeigt [113], was zweifellos die Verwendung von ETD in zukünftigen XL-MS-Studien erleichtern wird.
1.3 Anreicherung vernetzter Peptide zur Verbesserung ihrer Detektion
Eine weitere Herausforderung bei Vernetzungsstudien ist die geringe Häufigkeit von vernetzten Peptiden im Vergleich zu nicht-vernetzten Peptiden in komplexen Peptidgemischen. Dies liegt daran, dass Vernetzungsreaktionen keine homogenen Ereignisse sind und oft zu heterogenen vernetzten Spezies führen. Die Vernetzungseffizienz hängt von mehreren Faktoren ab, einschließlich Proteinkonzentration, Vernetzungschemie, Residuen-Proximity und Oberflächenzugänglichkeit. Darüber hinaus erschweren Variationen in Vernetzungsereignissen das Gemisch zusätzlich, was zu dead-end-modifizierten (Typ 0), intra-vernetzten (Typ 1) und inter-vernetzten (Typ 2) Peptiden führt [114]. Inter-vernetzte Peptide – hier als vernetzte Peptide bezeichnet – sind die strukturell informativsten Spezies, aber in der Regel auch die am wenigsten häufigen. Letztendlich nimmt die Heterogenität vernetzter Peptide exponentiell mit der Proteinkomplexität zu, was die Anreicherung vernetzter von linearen Spezies zu einer Notwendigkeit macht – insbesondere für proteomweite Studien –, um die Detektion gering-abundanter vernetzter Peptide während datenabhängiger Akquisitionen zu verbessern.
1.3.1 Chromatographische Trennung vernetzter Peptide
Unterschiede in den physikochemischen Eigenschaften von vernetzten im Vergleich zu linearen Peptiden wurden für die Peptid-Ebene-Trennung unter Verwendung chromatographischer Techniken genutzt. Da vernetzte Peptide typischerweise höhere Ladungen aufweisen und größer sind als lineare Peptide (einschließlich dead-end-modifizierter und intra-vernetzter Peptide), werden stark-Kationenaustausch- (SCX) [46] und Peptid-Größenausschlusschromatographie (SEC) [115, 116] häufig zur Anreicherung vernetzter Peptide eingesetzt. Obwohl beide Methoden effektiv sind, ist die SCX-Trennung flexibler und kann mit SCX-Tips wie StageTips [117] durchgeführt werden. Peptid-SEC bietet jedoch eine bessere Auflösung als SCX bei der Unterscheidung von vernetzten Peptiden von linearen Peptiden [115]. Um die SCX-Trennung vernetzter Peptide weiter zu verbessern, wurde kürzlich eine ladungsbasierte fraktionelle diagonale Chromatographie (XL-ChaFRADIC) [118] beschrieben, die eine zweidimensionale SCX-Trennung integriert. Die erste Dimension isoliert hochgeladene Spezies aus einem Lys-C-Verdau, die dann weiter durch Trypsin verdaut und einer zweiten SCX-Dimension unterzogen werden. Eine weitere aktuelle Studie verwendete eine Kombination aus Protein-Gel-Filtration, gekoppelt mit diagonaler Peptid-SCX, um die Identifizierung von Vernetzungen aus proteomweiten Gemischen zu erleichtern [119]. Obwohl eine solche Kombinationsstrategie die Komplexität des endgültigen Peptidgemisches tatsächlich reduziert, ist zu beachten, dass die mehrdimensionale Chromatographie auch zu Probenverlusten führen kann, insbesondere bei bereits gering-abundanten vernetzten Peptiden. Daher ist bei der Auswahl der Trennungsmethoden eine sorgfältige Abwägung erforderlich, um die Probenwiederfindung und die Identifizierung von Vernetzungen zu maximieren.
1.3.2 Affinitätsreinigung vernetzter Peptide
Eine weitere Strategie zur Verbesserung der Empfindlichkeit der XL-MS-Analyse ist die Anreicherung vernetzter Peptide durch Affinitätsreinigung. Dies kann durch das Design anreicherbarer Vernetzer erreicht werden, die eine Affinitätshandle enthalten. Unter den verschiedenen Affinitäts-Tags, die in der Proteinreinigung weit verbreitet sind, scheint der Biotin-Tag aufgrund seiner hohen Affinität zu Streptavidin (Kd~10−15) für die Peptidreinigung am beliebtesten zu sein. Im Laufe der Jahre wurden verschiedene Arten von Biotin-getaggten Vernetzern entwickelt, darunter nicht-spaltbare und MS-spaltbare Vernetzungsreagenzien [68, 75, 76, 120–124]. Obwohl es praktisch ist, Biotin-getaggte Vernetzungsreagenzien direkt zu verwenden, ist es vorteilhaft, affinitätsbasierte Vernetzer ohne den voluminösen Biotin-Tag zu entwerfen, um ihre geringe Größe, Zugänglichkeit und Membranpermeabilität zu erhalten. Alternativ kann der Biotin-Tag zu einem späteren Zeitpunkt durch die Verwendung von Azid- oder Alkin-getaggten Vernetzern mit einer „Click-Chemie“-basierten Konjugation [79, 93, 125–129] integriert werden. Nach der Anreicherung können biotinylierte vernetzte Peptide direkt von Affinitätsharzen eluiert werden. Die Entfernung des Biotin-Tags vor der MS-Analyse wird jedoch bevorzugt, da das Vorhandensein von Biotin die LC-MS-Analyse vernetzter Peptide erschweren kann [124]. Dies kann durch die Integration verschiedener chemischer Spaltstellen in die Linker erreicht werden, wie z.B. säure-spaltbare (z.B. Azid/Alkyne-A-DSBSO) [79, 93] und Azobenzol-basierte spaltbare Stellen (z.B. Leiker) [124].
Abgesehen von der Integration von Affinitätshandles in Vernetzern hat eine kürzlich durchgeführte Studie die Machbarkeit einer alternativen Strategie demonstriert, die auf der Fusion von Zielproteinen mit einem modifizierten His-Tag, d.h. CH (Cystein-Histidin)-Tag, basiert, der aus einem einzelnen Cystein, gefolgt von einer „DP“ (Asparaginsäure-Prolin)-Einheit und einem Histidin-Tag besteht [130]. Der Cysteinrest fungiert als Ankerstelle für heterobifunktionale Vernetzer, wobei ein Ende auf Cysteine abzielt, der His-Tag für die Affinitätsreinigung und die DP-Bindung zur Unterstützung der MS-Analyse. Diese Strategie zielt darauf ab, nur CH-Tag-enthaltende vernetzte Peptide zu erfassen, wodurch nur gezielte Proteininteraktionsregionen für die Analyse angereichert werden. Diese Arbeit deutet darauf hin, dass Affinitäts-Tags, die nicht Biotin-Tags sind, für die Affinitätsreinigung vernetzter Peptide in zukünftigen Studien erforscht werden könnten. Für die effiziente Verwaltung der generierten Datenmengen aus solchen Experimenten könnte eine gut strukturierte zeiterfassung excel einfach eine wichtige Rolle spielen.
2. Entwicklung neuer Vernetzungschemien
Die bisher am häufigsten verwendeten Vernetzer sind homobifunktionale, auf Amine (Lysin) abzielende Reagenzien, die aus N-Hydroxysuccinimidyl (NHS)-Ester-Funktionsgruppen bestehen (z.B. Disuccinimidylsuberat (DSS)). Das Anzielen von Lysinresten ist aus mehreren Gründen vorzuziehen: ihre relativ hohe Gesamtprävalenz (~6 % aller Reste), ihre Verteilung über lösungsmittelzugängliche Proteinoberflächen und die Spezifität primäramin-zielender Chemien. Das Anzielen von Lysinresten für die Vernetzung ist jedoch weniger geeignet für die Erfassung hydrophober Oberflächeninteraktionen, die vergraben sein und geladene Reste fehlen können. Ähnlich können Proteine mit wenigen oder keinen Lysinresten schwierig zu charakterisieren sein, wenn aminzielende Reagenzien verwendet werden. Im Laufe der Jahre wurden verschiedene Vernetzungsreagenzien und Workflows entwickelt, die jeweils einzigartige chemische Strukturen und Kombinationen von Funktionsgruppen aufweisen, um das Spektrum der XL-MS-Anwendungen zur Untersuchung von Proteinstrukturen zu diversifizieren.
2.1 Säurerestspezifische Vernetzer
Asparaginsäure- (Asp) und Glutaminsäure- (Glu) Reste machen ungefähr 12 % aller Aminosäurereste aus und besetzen häufig exponierte Oberflächenbereiche von Proteinen, die für Protein-Protein-Interaktionen von entscheidender Bedeutung sind. Daher stellen sie hochpotenzielle Vernetzungsziele zur Kartierung von Proteininteraktionskontakten dar. Frühere Arbeiten von Novak und Kruppa beschrieben die Machbarkeit der säurerestspezifischen Vernetzung bei ~pH 5,5 unter Verwendung von EDC-aktivierten Asp und Glu mit Dihydrazid-Vernetzer [131]. Dieser Workflow wurde später von Leitner et al. verbessert, die DMTMM als Kupplungsreagenz anstelle von EDC verwendeten, wodurch die Asp- und Glu-Vernetzung bei neutralem pH-Wert erfolgen konnte – besser geeignet für die Aufklärung von Proteinstrukturen unter physiologischen Bedingungen [132]. Unser Labor erweiterte diese Vernetzungschemie durch die Entwicklung eines sulfoxidhaltigen, MS-spaltbaren, säurerestspezifischen Dihydrazids (d.h. DHSO), das den gleichen MSn-Workflow verwendet, der für die Identifizierung von Lysin-reaktiven DSSO-vernetzten Peptiden entwickelt wurde (Abbildung 2A) [85]. Aufgrund des höheren Grades an Variabilität und Komplexität beim Vergleich von säurerestspezifischer mit lysin-zielender Vernetzung vereinfacht die MS-Spaltbarkeit von DHSO die Identifizierung von Asp/Glu-vernetzten Peptiden drastisch und liefert somit komplementäre Interaktionskontakte zur Erleichterung der Aufklärung von Architekturen von Proteinkomplexen.
2.2 Nicht-spezifische, photoaktivierte Vernetzer
Diazarin-inkorporierte Aminosäureanaloga unterscheiden sich von restspezifischen Vernetzern durch ihre Fähigkeit, unspezifisch proximale Reste zu vernetzen [18, 133, 134]. Diese Funktionalität hat einen Vorteil gegenüber ortsspezifischen Vernetzungsreagenzien, da die unspezifische photoinduzierbare Chemie die Vernetzung von Resten in hydrophoben Regionen ermöglicht und die Anwendung der Vernetzung auf die strukturelle Bestimmung von lösungsmittelunzugänglichen Interaktionsregionen, z.B. membrandurchspannende Proteinkomplexe [135], erweitert. Photoreaktive Funktionsgruppen (d.h. Diazarin, Phenylazid, Benzophenon) wurden auch in Verbindung mit NHS-Estern untersucht, um kurze, heterobifunktionale Vernetzungsreagenzien zu erzeugen, die lysin-zielende und unspezifische Enden enthalten [136, 137]. Obwohl die unspezifische Chemie attraktiv ist, um die Oberflächenabdeckung zu erhöhen, führt sie oft zu hochkomplexen vernetzten Produkten, was die Datenbanksuche erschwert. Darüber hinaus kann jede Interaktion durch mehrere Spezies vernetzter Peptide beschrieben werden, wodurch deren individuelle Häufigkeiten effektiv verdünnt werden. Zusammen stellen diese Probleme Herausforderungen für die empfindliche MS-Analyse und die genaue Identifizierung vernetzter Peptide dar, was ihre derzeitige Anwendung auf Studien einzelner Proteine begrenzt.
3. XL-MS-Strategien für die strukturelle Analyse von Proteinkomplexen
Die XL-MS-Technologie ist ein leistungsstarkes und effektives Strukturwerkzeug, da sie durch die Identifizierung vernetzter Peptide mehrere Informationsebenen bietet [4, 6, 7, 23, 38, 79, 138, 139]. Innerhalb eines gegebenen Proteinkomplexes identifiziert die Vernetzungsanalyse proximale Reste zwischen Untereinheiten von Proteinkomplexen und liefert Hinweise auf die räumliche Orientierung und Protein-Konnektivität. Zum Beispiel wurde die Vernetzung erfolgreich mit phylogenetischer Sequenzalignierung gekoppelt, um potenzielle evolutionär konservierte, funktionell wichtige Reste zu identifizieren. Nachfolgende Mutationsstudien von Kandidatenstellen bestätigten Funktionsverlust-Mutanten, was die Lokalisierung von Proteininteraktionsflächen in Schizosaccharomyces pombe-Telomerkomplexen ermöglichte [97, 100]. Ähnlich wurden Vernetzungsdaten verwendet, um Trunkierungs-/Deletionsmutagenese zur Identifizierung von Proteinbindungsdomänen, die an Exozytose und Mikrotubuli-Komplexen beteiligt sind, zu steuern [140]. Identifizierte physikalische Kontakte zwischen Proteinkomplexen können auch dazu beitragen, die Ränder von Proteininteraktionsnetzwerken zu definieren und experimentell abgeleitete Interaktionstopologien zu generieren, die normalerweise mehrere Runden reziproker Co-Immunpräzipitation in traditionellen massenspektrometrischen Ansätzen erfordern [38]. Darüber hinaus enthalten Vernetzungen definierte räumliche Restriktionen, die zur Bestätigung bestehender hochauflösender Strukturen und/oder zur Unterstützung der computergestützten Modellierung verwendet werden können. Neben Inter-Protein-Interaktionen werden auch Details über Intra-Protein-Interaktionen für jeden Proteinkomplexbestandteil erhalten, die weiter zur architektonischen Aufklärung multimerer Assemblies beitragen können. Angesichts dieser einzigartigen Fähigkeiten wurden XL-MS-Studien zur strukturellen Charakterisierung verschiedener Proteinsysteme eingesetzt [19, 28, 41, 42], die auf eine wegweisende Veröffentlichung im Jahr 2000 zurückgehen, in der Young et al. das Potenzial von XL-MS demonstrierten, indem sie Vernetzungsdaten mit computergestützter Modellierung koppelte, um die dreidimensionale Proteinfaltung eines Modellproteins FGF-2 vorherzusagen [37]. Im selben Jahr verwendeten Rappsilber und Mitarbeiter Vernetzung, Gelelektrophorese und Proteinidentifizierung, um die räumliche Organisation von Untereinheiten innerhalb eines Proteinkomplexes zu bestimmen, obwohl vernetzte Peptide nicht identifiziert wurden [141]. Trotz dieser frühen Arbeiten, die den Weg für XL-MS-Studien ebneten, wurden erfolgreiche Beispiele für die strukturelle Analyse von Proteinkomplexen erst fast ein Jahrzehnt später berichtet. Im Jahr 2010 kartierten Chen et al. die Interaktion zwischen RNA-Polymerase II und dem TFIIF-Komplex durch die Identifizierung von 352 Inter-Untereinheiten-Vernetungen, was die Lokalisierung der Pol II-TFIIF-Interaktionsfläche ermöglichte [32]. Seitdem haben sich die XL-MS-Anwendungen – insbesondere in den letzten Jahren – aufgrund dramatischer Fortschritte bei Bioinformatik-Tools und Vernetzungsreagenzien, wie oben beschrieben, erheblich weiterentwickelt. Darüber hinaus wurden verschiedene XL-MS-Strategien entwickelt, um die strukturelle Analyse von Proteinkomplexen zu erleichtern.
3.1 Kombinatorische Vernetzungsstrategien zur Gewinnung umfassender Informationen
Eines der langfristigen Ziele in XL-MS-Studien ist die Generierung von Daten, die für die de novo-Strukturmodellierung von Proteinen und Proteinkomplexen ausreichen. Es gibt jedoch zwei große Herausforderungen bei der de novo-Computergestützten Vorhersage: die Stichprobenziehung von Strukturen und die Diskriminierung ungenauer Strukturen [142]. Die Bewertung des Einflusses von XL-MS-Daten auf die de novo-Strukturvorhersage hat gezeigt, dass eine ausreichende Anzahl von Abstandsrestriktionen, die aus XL-MS-Daten generiert werden, nicht nur vorteilhaft ist, um die Größe des Stichprobenraums zu verringern, sondern auch um die Diskriminierungskraft der Bewertungsfunktion zu erhöhen und so die Identifizierung genauer Modelle zu verbessern [142].
Um die Ausbeute an strukturellen Daten, die durch XL-MS-Analysen gewonnen werden können, zu erhöhen, können verschiedene Vernetzungsreagenzien, die auf unterschiedliche Reste abzielen, eingesetzt werden, um umfassendere Informationen zu erhalten. Zum Beispiel zeigte die Chait-Gruppe, dass die Kombination von Lysin-reaktiven Reagenzien wie DSS (Amin-zu-Amin) mit dem Null-Längen-Vernetzer EDC (Amin-zu-Carboxyl) zu komplementären Strukturinformationen führte, wie in ihrer Charakterisierung des Nup84-Komplexes [36] belegt. Ähnliche Kombinationen wurden auch von anderen Laboren übernommen [17, 143, 144]. Darüber hinaus bietet die Integration von NHS-Estern und säurerestspezifischen Dihydraziden ebenfalls komplementäre Karten von Proteininteraktionen [85, 132]. Andere Kombinationen, wie Lysin-reaktive Reagenzien mit photoinduzierten unnatürlichen Aminosäuren, waren ebenfalls erfolgreich, aufgrund ihrer orthogonalen Fähigkeiten, lösungsmittelzugängliche bzw. lösungsmittelunzugängliche Regionen anzuzielen [145, 146]. Neuartige heterobifunktionale Vernetzer, die Lysin-zielende NHS-Chemie mit unspezifischen photoaktivierbaren Diazarenen (Sulfo-SDA, Sulfo-SBP) kombinieren, haben dazu beigetragen, die durch XL-MS gewinnbare Informationsmenge weiter zu erhöhen und „hochdichte“ Datensätze mit größerem Potenzial für die de novo-Modellierung zu liefern [137, 147]. Kollektiv hat die Integration verschiedener Vernetzungschemien ihre Wirksamkeit zur Erweiterung der Abdeckung von Interaktionskarten demonstriert.
Neben der Reste-zielenden Chemie kann auch die Länge des Vernetzers die Anzahl der erhältlichen vernetzten Peptide und ihren informativen Interaktionsgehalt beeinflussen [6, 132, 142]. Während kürzere Vernetzungsreagenzien oft zu weniger Vernetzungen führen, stellen ihre Übersetzung in räumliche Informationen “engere” Abstandsrestriktionen für die Strukturmodellierung dar. Längere Vernetzer hingegen liefern typischerweise höhere Anzahlen von Vernetzungen, die möglicherweise nicht so strukturell informativ sind, wodurch sie besser für Interaktionserfassungsstudien geeignet sind [142]. Da XL-MS die Erfassung aller Arten von Interaktionen (d.h. stabil, schwach, dynamisch/transient) in einem einzigen Experiment ermöglicht, stellen die resultierenden Vernetzungsdaten im Allgemeinen den Durchschnitt mehrerer struktureller Ensembles dar [148]. Daher ist es konzeptionell denkbar, dass die Integration von Vernetzungsreagenzien unterschiedlicher Längen die Tiefe der Strukturinformationen erhöhen kann, indem Proteininteraktionen innerhalb verschiedener Konformationen von Proteinkomplexen für eine genauere Strukturvorhersage erfasst werden [142]. Obwohl gezeigt wurde, dass unterschiedliche Linkerlängen komplementäre Ergebnisse liefern [132, 148, 149], bleibt unklar, wie viele zusätzliche wertvolle Informationen gewonnen werden können, um die computergestützte Modellierung zu verbessern. Häufig verwendete Vernetzer, die optimale Vernetzungslängen (10~15Å) aufweisen [6, 142], haben ihre Wirksamkeit bei der Aufklärung von Architekturen von Proteinkomplexen auf Systemebene bewiesen [38, 101, 102], während signifikant längere PIR-Vernetzer für die Kartierung von Proteininteraktionen gleichermaßen erfolgreich waren [40, 105–108, 139, 150, 151]. Es ist offensichtlich, dass weitere Untersuchungen zum Einfluss der Vernetzerlängen auf die strukturelle Charakterisierung notwendig sind, um XL-MS-Experimente zu optimieren.
3.2 Probenvorbereitung für die in vitro XL-MS-Analyse von Proteinkomplexen
3.2.1 In vitro On-Bead-Vernetungsstrategien
Die meisten bisherigen in vitro XL-MS-Studien an Proteinkomplexen basierten auf rekombinanten Proteinkomplexen als Quelle hochreiner und homogener Materialien für in-solution XL-MS-Experimente (Abbildung 3A). Entwicklungen in der Probenvorbereitung haben die Anwendung von XL-MS auf heterogenere Studien, wie affinitätsgereinigte Proteinkomplexe aus nativen Zellen, erweitert. Eine wegweisende Studie von Herzog et al. demonstrierte die Machbarkeit der Durchführung von On-Bead-Vernetung und Verdau von His-getaggten Proteinkomplexen, die aus menschlichen Zellen affinitätsgereinigt wurden (Abbildung 3B), was sowohl die Reaktionseffizienz als auch die Analyseempfindlichkeit erhöhte [38]. Infolgedessen ermöglichte das optimierte XL-MS-Protokoll eine systematische Analyse menschlicher Phosphatase-2A (PP2A)-Komplexe und die Generierung einer umfassenden Interaktionsnetzwerk-Topologie von PP2A-Komplexen, die 176 Inter-Protein- und 570 Intra-Protein-Vernetungen enthielten. Darüber hinaus lieferten die Vernetzungsdaten Abstandsrestriktionen zur Modellierung der Interaktionen zwischen PP2A-regulatorischen Proteinen mithilfe von ROSETTA.
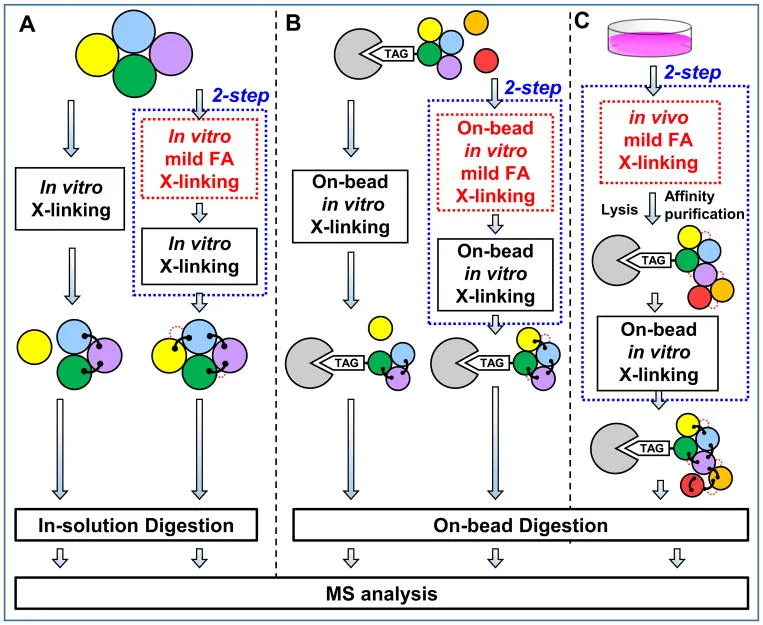 In vitro Vernetzungsstrategien für Proteinkomplexe.
In vitro Vernetzungsstrategien für Proteinkomplexe.
Zusätzlich zum His-Tag kann der Biotin-Tag für Reinigungen unter nativen und vollständig denaturierenden Bedingungen verwendet werden. Im Laufe der Jahre haben wir mehrere neue HB (d.h. Histidin-Biotin)-Tag-basierte Affinitätsstrategien entwickelt, um Proteinkomplexe aus menschlichen Zellen für XL-MS-Studien effektiv zu isolieren [79, 99, 152, 153]. Ähnlich haben wir auch On-Bead-Vernetung und -Verdau von HB-getaggten Proteasomkomplexen eingesetzt, was sich mit einer einstufigen Streptavidin-Reinigung als robust und effektiv erwies [98, 99]. Die extrem starke Bindungsaffinität zwischen Biotin und Streptavidin ermöglicht Reinigungen mit höherer Spezifität und weniger Hintergrund im Vergleich zur His-Tag-basierten Reinigung in Säugetierzellen. Es ist erwähnenswert, dass Streptavidin-Beads die XL-MS-Analyse nicht zu stören scheinen. Insgesamt haben unsere Ergebnisse die Vorteile der On-Bead-Verarbeitung von Proteinkomplexen für Vernetzungsstudien weiter demonstriert.
Im Allgemeinen wird die Antikörper-basierte Reinigung für XL-MS-Experimente nicht bevorzugt, da Antikörper empfindlich gegenüber Pufferbedingungen sind und die Peptidanalyse stören. Um diese Probleme zu umgehen, erstellte die Chait-Gruppe kürzlich Lysin-lose GFP-Nanobodies, um die Reinigung von GFP-getaggten Proteinkomplexen und die On-Bead-Vernetung mit Lysin-reaktiven Vernetzern vor der anschließenden SDS-PAGE und In-Gel-Verdau zu erleichtern [138]. Dieser Workflow wurde erfolgreich angewendet, um die Architekturen mehrerer Proteinkomplexe, z.B. Exosomen, zu definieren [138]. Kürzlich wurde gezeigt, dass ein kleiner, kommerziell erhältlicher GFP-Nanobody (12 kDa) auch für ähnliche XL-MS-Analysen verwendet werden kann [154]. Es sind jedoch umfangreiche Waschschritte und eine strenge Datenfilterung erforderlich, um störende Identifizierungen aufgrund von Nanobody-Interferenzen zu minimieren. Insgesamt scheint die On-Bead-Vernetung affinitätsgereinigter Proteinkomplexe (Abbildung 3B) attraktiver zu sein als die In-Solution-Vernetung, da sie eine flexible Probenhandhabung ermöglicht, um eine optimale Vernetzung von Proteinkomplexen bei niedrigen Konzentrationen mit minimalem Probenverlust zu gewährleisten.
3.2.2 Zwei-Schritt-Vernetungsprotokolle
Obwohl die meisten XL-MS-Studien an Proteinen und Proteinkomplexen effektive einstufige Reaktionen mit einem ausgewählten Vernetzer verwenden, werden möglicherweise nicht alle transienten Interaktionen ausreichend erfasst und charakterisiert. Um die Detektion schwacher Interaktionen zu verbessern und ausreichende vernetzte Produkte für die MS-Analyse zu generieren, wurde ein Zwei-Schritt-Vernetungsprotokoll erfolgreich angewendet, um die Dynamik von Mediator-RNA-Polymerase II Pre-Initiation Complexes zu analysieren (Abbildung 3A) [155]. Diese Strategie verwendete einen substöchiometrischen Formaldehyd (FA)-Fixierungsschritt als ersten Schritt, um Konfigurationsdynamiken einzufrieren, bevor die SBAT (d.h. 1-Hydroxy-7-azabenzotriazol-Analogon von DSS)-Vernetung erfolgte [155]. Dieser Fixierungsschritt schien die nachfolgende Vernetzung und Identifizierung vernetzter Peptide nicht zu stören, und die Ergebnisse deuten darauf hin, dass eine solche Stabilisierung die strukturelle Charakterisierung von Mediator-Polymerase-Komplexen durch XL-MS erheblich unterstützte. Darüber hinaus wurde auch gezeigt, dass SBAT eine schnellere lysinreaktive Chemie aufweist als DSS [155, 156]. Zusammen ermöglichte dieses überarbeitete Vernetzungsverfahren die architektonische Aufklärung des gesamten Komplexes und einzelner Unterkomplexe sowie ihrer strukturellen Dynamik [155].
Zusätzlich zur in vitro-Fixierung wurde die FA-Vernetung in vivo weit verbreitet zur Stabilisierung von Protein-Protein-Interaktionen in intakten Zellen vor der Zelllyse und Proteinreinigung unter nativen [98, 99, 157, 158] und denaturierenden Bedingungen [159–162] eingesetzt. Kürzlich haben wir zwei Affinitätsreinigungsstrategien entwickelt, nämlich XAP (in vivo Cross-Linking (X) Assisted Affinity Purification) [98] und XBAP (in vivo Cross-Linking (X) Assisted Bimolecular Tandem Affinity Purification) [153], um dynamische Interaktoren von Proteinkomplexen bzw. Unterkomplexen zu untersuchen. Diese Ansätze verwenden eine milde in vivo FA-Vernetung (<0,1%), die eine bessere Erhaltung dynamischer Interaktionen für die Affinitätsreinigung unter nativen Bedingungen ermöglicht. Darüber hinaus haben wir gezeigt, dass mit der XAP-Methode gereinigte HB-getaggte Proteasomkomplexe direkt einer in vitro On-Bead DSSO-Vernetung und LC-MSn-Analyse unterzogen werden können, wodurch wir die interaktionsvermittelte Regulation von Proteasomen untersuchen können [98]. Dieses Zwei-Schritt-Protokoll kombiniert eine milde in vivo FA-Fixierung vor der in vitro DSSO-Vernetung (siehe Abbildung 3C und Abschnitt 3.2.2) und ist vorteilhaft für die Untersuchung affinitätsgereinigter Komplexe aufgrund seiner Fähigkeit, schwache, transiente und/oder dynamische Interaktionen zu erhalten. Abgesehen von FA wurde auch die Glutaraldehyd-Vernetung zur Stabilisierung von Proteininteraktionen nach dem Kryofräsen von schockgefrorenen Zellen vor der Affinitätsreinigung eingesetzt [163]. Potenziell kann eine solche Vorbehandlung auch in Zwei-Schritt-Vernetungsverfahren integriert werden, wenn Zellen bei niedriger Temperatur verarbeitet werden. Es ist klar, dass Zwei-Schritt-Sequenzierungsverfahren bestimmte Vorteile für in vitro XL-MS-Studien bieten, insbesondere zur Erhaltung dynamischer/schwacher Interaktionen. Systematische Vergleiche zum besseren Verständnis der Auswirkungen der Vorfixierung würden jedoch zweifellos zur Optimierung zukünftiger experimenteller Protokolle beitragen.
3.2.3 In vivo-Vernetung von Proteinkomplexen
In den letzten Jahren haben in vitro XL-MS-Studien an Proteinkomplexen enorme molekulare Details zum Verständnis ihrer Architekturen, Funktionen und Regulation geliefert [11, 12, 19, 27, 28, 36, 40–42, 155, 164, 165]. Es ist jedoch biologisch relevanter, in vivo-Vernetungsexperimente durchzuführen, da diese eine authentischere Karte von Protein-Protein-Interaktionen (PPIs) liefern können, die in nativen zellulären Umgebungen auftreten [30]. Dies liegt daran, dass PPIs in Zellen hochdynamisch sind und durch verschiedene zelluläre Signale, einschließlich posttranslationaler Modifikationen, moduliert werden können. Während stabile Interaktionen verschiedene Reinigungsbedingungen überleben können, gehen transiente und schwach interagierende Proteine oft während des Prozesses verloren. Darüber hinaus kann eine native Zelllyse zu einer Reorganisation von PPIs führen und nicht-physiologische Interaktionen ergeben. Um einen wahren Schnappschuss von Proteininteraktionsnetzwerken in lebenden Zellen zu erhalten, ist es daher unerlässlich, diese Proteininteraktionen vor der Zelllyse zu stabilisieren/einzufrieren, was durch in vivo-Vernetung erreicht werden kann, eine einzigartige Fähigkeit, die andere Strukturwerkzeuge nicht besitzen. Bisher wurden nur eine begrenzte Anzahl von in vivo XL-MS-Studien berichtet, hauptsächlich aufgrund der oben diskutierten Herausforderungen. Während das Bruce-Labor sich auf die in vivo-Vernetung auf Proteomebene unter Verwendung seiner PIR-Vernetzer konzentriert hat [40, 77, 89, 104–106, 108, 139, 151], hat sich unser Labor hauptsächlich auf die in vivo XL-MS-Analyse von Proteinkomplexen konzentriert [79, 99, 159, 162, 166, 167]. Kürzlich haben wir einen membranpermeablen, sulfoxidhaltigen MS-spaltbaren und anreicherbaren NHS-Ester-Vernetzer Azide-A-DSBSO entwickelt (Tabelle 2) [79]. Dieser multifunktionale Vernetzer besitzt mehrere einzigartige Merkmale (d.h. geringe Größe, geeignete Spacerlänge (~14 Å), bio-orthogonaler Affinitäts-Tag, robuste MS-spaltbare Bindungen und eine Säurespaltstelle), was ihn zu einem attraktiven Reagenz zur Definition von PPIs in Zellen macht. Für die Kartierung der Interaktionsnetzwerktopologie von Proteinkomplexen in Säugetierzellen wird zunächst die in vivo Azide-A-DSBSO-Vernetung intakter Zellen durchgeführt, gefolgt von einer HB-Tag-basierten Tandem-Affinitätsreinigung [159], um in vivo-vernetzte Proteinkomplexe unter vollständig denaturierenden Bedingungen (d.h. 8M Harnstoff) zu extrahieren und nicht-spezifischen Hintergrund zu eliminieren. Vernetzungen werden dann durch Click-Chemie-basierte Konjugation biotinyliert, bevor die Proteinverdauung erfolgt, gefolgt von einer anschließenden Peptid-Affinitätsanreicherung. Vernetzte Peptide werden über die Säurespaltstelle eluiert, während der Biotin-Tag-enthaltende Teil an das Harz gebunden bleibt. Die Zugabe und Entfernung des Biotin-Tags eliminiert dessen Intervention während der Vernetzung bzw. MS-Analyse. Diese Strategie wurde erfolgreich auf die Untersuchung von Proteasomkomplexen angewendet und identifizierte erstmals in vivo Inter- und Intra-Untereinheiten-Proteasominteraktionen [79]. Darüber hinaus ist Azide-A-DSBSO in der Lage, in vivo PPIs auf Proteomebene zu analysieren [79]. Kürzlich haben wir auch die Machbarkeit der Verwendung von DSSO für die in vivo-Vernetung in Säugetierzellen [99] in Verbindung mit der HB-Tag-basierten Reinigung von vernetzten Proteasomkomplexen untersucht. Durch diese Studien konnten wir umfangreiche Interaktionen innerhalb von Proteasom-Untereinheiten unter Verwendung verschiedener Zelllinien und fortgeschrittener Orbitrap-Instrumente identifizieren. Dennoch ist Azide-A-DSBSO aufgrund seiner Anreicherungskapazität, die für proteomweite Studien entscheidend ist, besser für in vivo-Vernetungsstudien geeignet.
4. Aufklärung der Architekturen von Proteinkomplexen
Obwohl die durch XL-MS-Studien bereitgestellten Restriktionen eine zentrale Rolle bei der Untersuchung der Topologien verschiedener Proteinkomplexe gespielt haben [19, 28, 41, 42], reichen Vernetzungsdaten allein typischerweise nicht aus, um Proteinkomplexe strukturell abzuleiten. Dies ist immer noch größtenteils auf die begrenzten Strukturinformationen zurückzuführen, die durch Vernetzung aufgrund der Restspezifität der Reagenzien und der jeweiligen Restverfügbarkeit in Proteinen gewonnen werden können, trotz der jüngsten Fortschritte in den Vernetzungsmethoden. Darüber hinaus stellen Vernetzungsdaten typischerweise einen Datensatz dar, der den “Durchschnittszustand” von Proteinen beschreibt, der mehrere Konformationen umfassen kann. Software wie XL-MOD wurde für die Analyse widersprüchlicher, hochzuverlässiger Vernetzungen entwickelt [168, 169], während DynaXL ein weiteres Beispiel für Software ist, die entwickelt wurde, um widersprüchliche Vernetzungsdaten zu interpretieren, indem angeregte Zustandsstrukturen modelliert werden, die aus abstandsverletzenden Vernetzungen resultieren können [170]. Ohne andere Informationsquellen bleibt die Übersetzung spärlicher Vernetzungsdaten in definitive Strukturen und Modelle jedoch schwierig. Die Kombination von Restriktionen aus XL-MS-Experimenten mit komplementären Daten aus anderen Strukturtechniken hat sich als am effektivsten erwiesen, um verschiedene Aspekte der Proteinstruktur zu untersuchen.
4.1 Kopplung von XL-MS mit anderen MS-basierten Strukturwerkzeugen
Die kombinatorische Bottom-up-Analyse vernetzter Proteine und die Top-down-Analyse mittels nativer/IM-MS haben sich als effektive Mittel zur Bestimmung der dreidimensionalen Strukturen von Multiprotein-Assemblierungen erwiesen [171–175]. Während native Massenspektrometrie-Ansätze in der Lage sind, die Gesamtkomplextopologie und die Untereinheitenstöchiometrie zu bestimmen, sind sie in der Regel nicht in der Lage, die Untereinheitenkonnektivität zu bestimmen und Interaktionsflächen abzubilden. Andererseits eignen sich XL-MS-Techniken gut zur Bestimmung der topologischenen Anordnung von Proteinkomplexen, zur Identifizierung von Proteininteraktionsregionen und zur Erhaltung dynamischer Interaktionen. Es ist klar, dass die beiden MS-basierten Ansätze einzigartige und komplementäre Strukturinformationen liefern können, die durch keinen der beiden allein leicht zugänglich wären. Daher bleibt die Kombination dieser beiden Strategien für die Strukturaufklärung makromolekularer Komplexe attraktiv [144, 146, 176, 177], obwohl die Anwendung aufgrund der Schwierigkeit, endogene Proteinassemblierungen in ausreichend hoher Konzentration und Reinheit für die native MS-Analyse zu extrahieren, leicht eingeschränkt ist.
XL-MS wurde auch zusammen mit oberflächenprobenden Methoden wie radiolytischem Footprinting [178, 179], kovalenter Markierung [176, 180, 181], HDX (Wasserstoff-Deuterium-Austausch) [182–184] und begrenzter Proteolyse [185, 186] eingesetzt, um die Strukturen von Proteinen sowie deren Dynamik und Konformationsänderungen zu untersuchen. Die Grundlage dieser Techniken liegt in ihrer Fähigkeit, die lösungsmittelzugänglichen Oberflächen von Proteinen zu identifizieren, die für Proteinfaltungsstudien und die Identifizierung lösungsmittelunzugänglicher Kontaktflächen interpretiert werden können. Footprinting- und Markierungsmethoden modifizieren lösungsmittel-exponierte Aminosäurerest-Seitenketten durch Oxidation oder chemische Modifikationen, die durch Standardproteomik identifiziert werden können, während HDX den In-Lösung-Austausch von Protonen gegen Deuterium nutzt. Begrenzte oder partielle Proteolyse-Methoden sind wahrscheinlich die am wenigsten empfindlichen, da sie auf kurze Inkubationen mit enzymatischen Proteasen angewiesen sind, um exponierte Schleifen und flexible Regionen zu bestimmen. Folglich können diese Techniken auch verwendet werden, um die Strukturkonformationen mehrerer (z.B. Wildtyp, Liganden-gebunden, PTM-modifiziert, PPI) Proteinzustände basierend auf ihren differentiellen Oberflächendynamiken zu vergleichen und gegenüberzustellen. Verschiedene Tools wurden entwickelt, um die Integration von räumlichen Restriktionen aus XL-MS mit anderen MS-basierten Strukturmethoden zu erleichtern (siehe Tabelle 1), zum Beispiel mit kovalenter Markierung zur Untersuchung lösungsmittelzugänglicher Oberflächen für die Proteinmodellierung [64, 180].
4.2 Integratives Modellieren makromolekularer Maschinen
Als Ergebnis eines jüngsten Anstiegs der Instrumentierung und der Bildinterpretationssoftware hat sich die Kryo-EM als alternative Methode zur strukturellen Aufklärung makromolekularer Assemblierungen mit (nahezu) atomarer Auflösung entwickelt. Im Vergleich zur Röntgenkristallographie und NMR ist der Probenpräparationsprozess für die Kryo-EM deutlich weniger aufwendig. Die Analyse konformationell oder zusammensetzungsmäßig heterogener Komplexe stellt jedoch immer noch eine Herausforderung dar, da es aufgrund der begrenzten Auflösung in EM-Karten oft Bereiche mit nicht zugewiesenen Elektronendichten gibt. Infolgedessen kann die genaue Lokalisierung einzelner Untereinheiten innerhalb eines Proteinkomplexes ohne andere Formen von Strukturinformationen schwierig sein. In diesen Szenarien können XL-MS-Daten orthogonale Informationen zu niedrigauflösenden EM-Dichtekarten liefern, um die spezielle Organisation einzelner Untereinheiten zu leiten [187, 188].
Letztendlich bleiben die meisten Proteinkomplexe gegenüber jeder einzelnen Strukturmethode unzugänglich, was zu einer Ära führt, die von kombinierten Workflows dominiert wird, die Computerprogramme zur Konsolidierung verschiedener Formen komplementärer Strukturinformationen nutzen (Abbildung 4). Von hochauflösenden partiellen Strukturen, die durch Röntgenkristallographie/NMR beigesteuert wurden, und niedrigauflösenden Dichtekarten aus Kryo-EM/SAXS-Studien bis hin zu Strukturinformationen aus XL-MS und anderen strukturellen MS-Strategien wurden “hybride” Plattformen wie IMP (Integrative Modeling Platform) [189, 190] entwickelt, um diverse Formen biophysikalischer und proteomischer Daten zur Strukturbestimmung zu integrieren. Daten aus verschiedenen Strukturmethoden werden in einen Satz von räumlichen Restriktionen übersetzt, die kumulativ eine Bewertungsfunktion beschreiben. Ausgehend von anfänglichen Zufallskonfigurationen wird der Raum der Konformationen iterativ untersucht, um die Bewertungsfunktion zu minimieren, was zu einem optimierten Ensemble führt, das die Erfüllung der ursprünglichen Daten maximiert. Wie in Tabelle 1 zusammengefasst, wurden Xlink Analyzer [191], ROSETTA [192] und HADDOCK [193] ebenfalls erfolgreich für ähnliche Studien eingesetzt, was den Weg für einen neuen Archetyp in der Strukturbiologie in Form des “integrativen Strukturmodellierens” ebnet. XL-MS war in den letzten Jahren maßgeblich an der integrativen Modellierung einer Reihe von Multiprotein-Assemblierungen beteiligt. Bemerkenswerte Beispiele sind der SAGA-Transkriptions-Co-Aktivatorkomplex [194–196], Elongator [197, 198], GPCRs [145, 199], ribosomale Komplexe [35, 164, 188] und der mitochondriale Komplex I [200]. Drei weitere makromolekulare Assemblierungen, die mit integrativen Ansätzen umfassend untersucht wurden, sind das 26S-Proteasom, der nukleäre Porenkomplex (NPC) und RNA-Polymerase-Komplexe, wie kürzlich in einer Übersichtsarbeit beschrieben [23]. Diese Komplexe wurden jedoch in den letzten Jahren kontinuierlich untersucht und haben weitere komplizierte Details zu ihrer Struktur und Funktion offenbart, die erwähnenswert sind. Darüber hinaus können die für die Untersuchung dieser Komplexe verwendeten Strategien auf andere Proteinkomplexe angewendet werden.
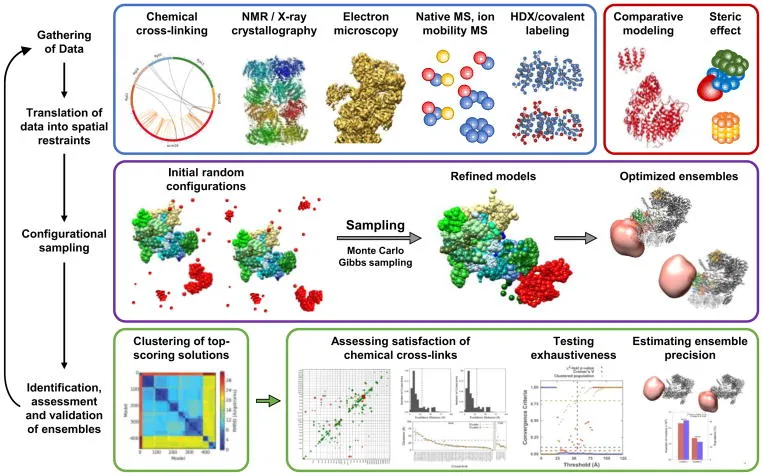 Allgemeines Schema für die integrative Strukturmodellierung.
Allgemeines Schema für die integrative Strukturmodellierung.
4.2.1 26S-Proteasom
Das 26S-Proteasom ist die makromolekulare Maschine, die für den kontrollierten Abbau ubiquitinierter Substrate verantwortlich ist und aus 33 Untereinheiten besteht, die zwei Unterkomplexe bilden: den 20S katalytischen Kernpartikel (CP) und den 19S regulatorischen Partikel (RP) [201]. Während das 20S CP für proteolytische Aktivitäten verantwortlich ist, unterstützt das 19S RP den Proteinabbau durch verschiedene Funktionen, einschließlich Substraterkennung, Protein-Deubiquitinierung, Entfaltung und 20S-Gating. Im Vergleich zum 20S CP ist das 19S RP viel dynamischer und flexibler – und kann weiter in die Basis- und Lid-Unterkomplexe unterteilt werden. Aufgrund seiner kompositionellen und konformationellen Heterogenität und dynamischen Natur war die strukturelle Analyse des 26S-Proteasom-Holokomplexes jahrzehntelang eine Herausforderung. Mit dem Fortschritt der Kryo-EM und hybriden Strukturwerkzeugen wurden in den letzten Jahren hochauflösende Strukturen von 26S-Proteasomen bestimmt [33, 202–205]. Dabei hat die XL-MS-Analyse maßgeblich zur Aufklärung der Proteasom-Architekturen und zur Entdeckung struktureller Details beigetragen, die der Proteasomfunktion und -regulation zugrunde liegen [23, 33, 95, 98, 99, 206–209]. In einer frühen Studie wurden XL-MS und EM verwendet, um die strukturelle Topologie des AAA-ATPase-Moduls im S. pombe 19S RP sowie dessen räumliche Beziehung zum α-Ring des 20S-Kernpartikels zu definieren [206]. Zur Charakterisierung der Architektur des Hefe-26S-Proteasoms verwendeten Lasker et al. ein integratives Strukturmodell, das Strukturinformationen aus XL-MS, EM, Röntgenkristallographie und Literatur nutzte [33]. Dieser kombinatorische Ansatz ermöglichte die Bestimmung der topologischen Anordnung und Form des 19S-Lids sowie dessen Interaktion mit dem 20S CP und der AAA-ATPase [33]. Diese Ergebnisse korrelierten sehr gut mit der Architektur des vollständigen 19S-Lids aus Hefe, das nur mittels EM und einem heterologen Expressionssystem erhalten wurde [203]. Obwohl die spätere Studie keine Versuche unternahm, Cross-Links zu identifizieren, wurde ortsspezifisches Cross-Linking durchgeführt, um in EM-Karten beobachtete Interaktionen zu validieren [203]. Zufälligerweise haben wir auch die topologische Anordnung der Hefe-19S-Basis- und Lid-Unterkomplexe basierend auf DSSO-Cross-Link-Daten und probabilistischer Modellierung abgegrenzt [95], wobei die gleiche räumliche Organisation wie berichtet [203] erhalten wurde. Darüber hinaus ergänzten unsere Cross-Link-Daten frühere Berichte, indem sie physikalische Kontakte zwischen den Untereinheiten Rpn12, Rpn13 und Rpn15 innerhalb des 26S-Proteasoms lieferten, wodurch deren Interaktionen weiter bestätigt und ihre Lokalisierung in den endgültigen Modellen unterstützt wurde. Dies war wichtig, da die geringe Größe von Rpn15 seine Zuordnung in EM-Karten verhindert hatte [33, 203, 206]. Zusätzlich zur Strukturbestimmung wurde ein integrierter Ansatz, der biochemische, EM- und XL-MS-Analysen kombiniert, erfolgreich zur Untersuchung der 20S-Biogenese eingesetzt, indem die Untereinheitenanordnung und Chaperonlokalisierung in proteasomalen Vorläufern untersucht wurden [207]. Die Ergebnisse haben gezeigt, dass das Hefe-Pba1-Pba2-Heterodimer während der 20S-Reifung aufgrund erheblicher Konformationsänderungen innerhalb der Alpha- und Beta-Ringe des 20S CP recycelt wird. Interessanterweise unterscheidet sich das Schicksal des Hefe-Pba1-Pba2-Heterodimers von seinem menschlichen Gegenstück PAC1-PAC2, das nach Abschluss der 20S-Assemblierung abgebaut wird [210]. Eine ähnliche integrierte Strategie wurde auch zur Analyse der 19S RP-Biogenese eingesetzt [209], die quantitative XL-MS für die vergleichende Analyse der Strukturen des 19S-Lid-Intermediats und des vollständigen Lids implementierte. Die Ergebnisse haben eine umfangreiche Umgestaltung des 19S-Lid-Vorläufers gezeigt, die durch Rpn12 vermittelt wird und so die RP-Assemblierung erleichtert. Protein-Protein-Interaktionen sind nicht nur für die Proteasom-Assemblierung und -Struktur, sondern auch für die Proteasom-Regulation von entscheidender Bedeutung. Ubp6 (Usp14 bei Säugetieren) ist eine Proteasom-gebundene Deubiquitinase, die wichtig für die Modulation des proteasomalen Abbaus ist [201]. Die strukturelle Analyse des Ubp6-26S-Proteasom-Komplexes mittels EM und XL-MS hat gezeigt, dass die aktive Stelle von Ubp6 zur Konformationslandschaft des 26S-Proteasoms beitragen kann [208].
Im Vergleich zu den Hefe-Proteasomen waren die Berichte über das menschliche 26S-Proteasom spärlich. Erst sehr kürzlich wurden zwei hochauflösende Kryo-EM-Strukturen (3,9 und 3,5 Å) des menschlichen 26S-Proteasoms beschrieben [204, 205]. Obwohl die Gesamtarchitektur des 26S-Holokomplexes von Hefe bis Mensch hochkonserviert ist, gibt es Unterschiede in den Konformationszuständen, der Untereinheitenzuordnung und der strukturellen Dynamik. Um die dynamischen Strukturen und die Regulation des menschlichen 26S-Proteasoms weiter zu verstehen, haben wir DSSO-basierte in vivo und in vitro XL-MS-Workflows eingesetzt, um PPIs innerhalb des 26S-Proteasoms umfassend zu untersuchen [99]. Als Ergebnis haben wir den größten Cross-Link-Datensatz für Proteasomkomplexe erhalten, mit 447 einzigartigen Lysin-zu-Lysin-Verknüpfungen, die 67 Inter-Protein- und 26 Intra-Protein-Interaktionen abgrenzen. In Kombination mit EM-Karten und computergestützter Modellierung haben wir Architekturen des menschlichen 26S-Proteasoms mit globalen RMSDs ≤ 1,3 mit berichteten hochauflösenden Strukturen [204, 205] aufgelöst, was minimale strukturelle Unterschiede anzeigt. Wichtig ist, dass die strukturelle Dynamik der Untereinheiten Rpn1, Rpn6 und Rpt6 basierend auf weniger aufgelösten Strukturen und mehreren Formen von Rpn1 und Rpn6, die in EM-Karten detektiert wurden, sowie einer großen Anzahl von abstandsverletzenden Cross-Links (>35 Å) für Rpn1 und Rpt6 abgeleitet wurde [99]. Diese Ergebnisse deuten auf eine konformationelle Heterogenität des menschlichen 26S-Proteasoms hin. Abgesehen vom 26S-Holokomplex wurden zusätzliche Cross-Links identifiziert, um direkte physikalische Interaktionen zwischen Proteasom-Untereinheiten und 15 Proteasom-interagierenden Proteinen zu demonstrieren, darunter 9 bekannte und 6 neue Interaktoren [99]. Unter ihnen ist UBLCP1 die einzige bekannte Proteasom-Phosphatase [211], deren dynamische Interaktion mit Rpn1 durch XL-MS und integrative Modellierung mit einem vorgeschlagenen Modell zur Ableitung der Wirkungsweisen von UBLCP1 bei der Regulierung von Proteasomen bestätigt wurde. Darüber hinaus legt die Validierung der ausgewählten PPIs mittels reziproker Reinigung und XL-MS nahe, dass Cross-Links als direkter Beweis für paarweise Proteininteraktionen dienen können, ohne weitere biochemische Bestätigung – wie oft für konventionelle AP-MS-Experimente erforderlich. Zudem haben wir gezeigt, dass die Kombination von in vivo und in vitro XL-MS-Analysen umfassendere Interaktionsdaten für die strukturelle Analyse von Proteasomkomplexen geliefert hat. Während in vitro-Studien reichlich Cross-Links für stabile Interaktionen liefern, sind in vivo-Experimente vorteilhaft für die Charakterisierung dynamischer und transienter Interaktionen.
Oxidativer Stress löst bekanntermaßen die Disassemblierung des eukaryotischen 26S-Proteasoms aus, wodurch mehr freies 20S CP zur Entfernung oxidierter Proteine freigesetzt wird [99, 212]. Das Proteasom-interagierende Protein Ecm29 wurde als Schlüsselregulator identifiziert, der die 26S-Proteasom-Disassemblierung moduliert. Um zu bestimmen, wie Ecm29 das 26S-Proteasom reguliert, haben wir XL-MS eingesetzt, um ihre Interaktionskontakte zu identifizieren. Da die Ecm29-Proteasom-Interaktion schwach und empfindlich gegenüber zellulären/experimentellen Bedingungen ist, haben wir XAP mit DSSO-basierter in vitro XL-MS (d.h. zweistufige Vernetzung (siehe Abbildung 3B und Abschnitt 3.2.2)) gekoppelt, um ausreichende Ecm29-Proteasom-Komplexe für die Strukturanalyse zu erhalten. Insgesamt wurden entsprechende Rest-Rest-Interaktionen zwischen Ecm29 und fünf 19S-Basiseinheiten, Rpt1, Rpt4, Rpt5, Rpn1 und Rpn10, identifiziert. Mittels integrativer Strukturmodellierung durch IMP wurde die Architektur des Ecm29-Proteasom-Komplexes bestimmt, zusammen mit einem vorgeschlagenen Modell für die Ecm29-abhängige Dissoziation des 26S-Proteasoms als Reaktion auf oxidativen Stress [98].
4.2.2 Nukleärer Porenkomplex
Der nukleäre Porenkomplex ist eine massive 50 MDa nukleozytoplasmatische Transportbaugruppe, die aus ungefähr 30 verschiedenen Nukleoporinen besteht, mit insgesamt mindestens 456 einzelnen Proteinen. Das Modell des Saccharomyces cerevisiae NPC war das erste, das durch einen integrativen Modellierungsansatz entwickelt wurde – es beschreibt die NPC-Architektur als eine hochkonservierte, symmetrische Organisation verschiedener, multisubunitärer Module [31]. In den folgenden Jahren wurden mehrere integrative Studien unter Verwendung von XL-MS eingesetzt, um diese einzelnen Module zu charakterisieren. Eine Studie des Beck-Labors, die Elektronen-Tomographie, Einzelpartikel-Elektronenmikroskopie und Vernetzungs-Massenspektrometrie nutzte, bestimmte die Struktur des menschlichen Nup107-Subkomplexes, eines wichtigen Gerüstmotivs des NPC, das über 32 Kopien zwei retikulierte Ringe überspannt [34]. Unter Verwendung einer Kombination aus Röntgenkristallographie, EM und Multi-Linker-Vernetung schlug das Chait-Labor ein integratives Modell für den Nup84-Subkomplex der Knospungshefe vor, eine hetero-heptamere Assemblierung, die den äußeren Ring des NPC über 16 stabile Kopien bildet [36]. Eine Veröffentlichung über den Nup84-Komplex aus Chaetomium thermophilum unter Verwendung von Kryo-EM und XL-MS zeigte ein dimeres Modul, das dem orthologen Nup107-Nup160-Komplex in höheren Eukaryoten ähnlicher ist [213], während weitere Forschungen zum NPC der Knospungshefe durch die Sali- und Rout-Gruppen ein multistabiles Modell für Nup133 unter Verwendung einer Kombination aus Röntgenkristallographiedaten, SAXS und Negativfärbe-EM, validiert durch Vernetzungsrestriktionen, aufzeigten [214]. Kürzlich wurde die Struktur des Nup82-Nup84-Komplexes unter Verwendung von Multi-Linker-Vernetung, EM, Röntgenkristallographie und SAXS gelöst [215]. Die Integration von Kryo-Elektronen-Tomographiedaten, Homologiemodellierung und der Anpassung hochauflösender Strukturen und Vernetzungsrestriktionen wurde verwendet, um eine umfassende Architektur für den menschlichen NPC zu bestimmen [216]. Die Konvergenz zwischen solchen aufgeklärten Strukturen und der biologischen Funktion des nukleären Porenkomplexes wurde sehr kürzlich in einer Übersichtsarbeit beleuchtet, die Aufschluss über die molekularen Mechanismen des nukleozytoplasmatischen Transports gibt [217].
4.2.3 RNA-Polymerase-Komplexe
Die RNA-Polymerase bildet zusammen mit den allgemeinen Transkriptionsfaktoren (GTFs), die den Präinitiationskomplex (PIC) und den Multiprotein-Mediator-Komplex umfassen, den grundlegenden eukaryotischen Transkriptionsapparat. Neben Pol II-TFIIF [32] wurden XL-MS-Studien zur strukturellen Aufklärung einzelner RNA-Polymerasen Pol I [218, 219], II [76], III [220] sowie PIC-gebundener Pol II [221, 222] und des Pol II-Capping-Komplexes [223] eingesetzt. Zusätzlich wurden die Strukturen des Mediator-Middle-Moduls [224] und des an RNA Pol II gebundenen Head-Moduls [225] ebenfalls mittels XL-MS untersucht. In den letzten Jahren wurden integrative Studien erweitert, um die komplexen Architekturen der RNA-Polymerase im Komplex mit GTFs [226, 227] und dem Mediator-Komplex [165, 228] zu untersuchen, was in der strukturellen Bestimmung des vollständigen Mediator-PIC gipfelte [155]. Diese Struktur berichtete über einen zuvor unbestimmten Proteinkinasekomplex, der TFIIK und die Mediator-Aktivator-Interaktionsregion der Med-PIC-Struktur involviert und den molekularen Mechanismus der Transkriptionsregulation durch Mediator aufdeckte. Andere interessante Studien im letzten Jahr berichteten auch über strukturelle Einblicke in die Initiation der Hefe-RNA-Polymerase I [229] und die molekulare Dynamik viraler Polymerasen, die die Annahme mehrerer funktionaler Konfigurationen ermöglichen [230].
5. Proteomweite XL-MS-Studien
Während die strukturelle Aufklärung isolierter Assemblierungen zweifellos entscheidend für ihr funktionelles Verständnis ist, stellt das Zusammenspiel zwischen diesen Proteinkomplexen und ihren Regulatoren, die für die homöostatische Aufrechterhaltung erforderlich sind, einen Forschungszweig dar, der erst begonnen hat, erschlossen zu werden. Daher besteht ein dringender Bedarf an einer systematischen, proteomweiten Charakterisierung von PPIs, um die Rollen und Funktionen von Proteinen und Proteinkomplexen vollständig zu verstehen. Verschiedene Studien haben das Potenzial von Vernetzungsmethoden gezeigt, nicht nur strukturelle Details endogener Interaktionen zu liefern, sondern auch einen Einblick in die umfassende Interaktionsprofilierung ganzer Proteome zu geben.
5.1 In vitro proteomweite Studien
Die Vereinfachung der MS-Analyse zur Identifizierung vernetzter Peptide, die durch MS-spaltbare Reagenzien ermöglicht wird, war besonders effektiv bei der Erhöhung der Ausbeute an XL-MS-Daten aus hochheterogenen Gemischen wie Zelllysaten. Frühe in vitro-Studien auf Proteomebene unter Verwendung nicht-spaltbarer Vernetzer lieferten nur begrenzte Informationen, selbst wenn sie mit Offline-Trennungstechniken gekoppelt wurden. Mehrere Versuche zur E. coli-Lysat-Vernetung wurden durchgeführt, wobei die ersten beiden jeweils weniger als hundert Vernetzungen ergaben und eine dritte 394 Vernetzungen identifizierte [44, 231, 232]. Die Studie von Yang et al., die die meisten Daten aus der E. coli-Lysat-Vernetung beisteuerte, berichtete auch 39 Vernetzungen aus den komplexeren C. elegans-Lysaten. Im Vergleich dazu wurden MS-spaltbare Vernetzer erfolgreich für groß angelegte Studien verschiedener Organismen eingesetzt. Zum Beispiel wurde BAMG effektiv zur Profilierung von HeLa-Zellkernextrakten in Verbindung mit diagonaler SCX-Trennung vernetzter Peptide eingesetzt [233], wobei 247 Vernetzungen bei einer geschätzten FDR von 0,4 % identifiziert wurden. Die Heck-Gruppe demonstrierte auch die Machbarkeit der Verwendung von DSSO und SCX-Fraktionierung für die proteomweite Analyse von HeLa- und Escherichia coli-Zelllysaten [101, 102], wobei 3.301 bzw. 1.158 einzigartige Vernetzungen bei 1 % FDR identifiziert wurden. Diese Studien legen nahe, dass MS-labile Vernetzungsreagenzien in Verbindung mit der Offline-Peptidanreicherung für in vitro XL-MS-Studien auf Proteomebene effektiver sind.
Kürzlich wurden die von Tan et al. entwickelten Leiker-Reagenzien für Vernetzungsstudien an Lysaten von E. coli und Caenorhabditis elegans eingesetzt [124]. Obwohl Leiker-Vernetzer nicht MS-labil sind, handelt es sich um biotinylierte Reagenzien, die eine selektive Anreicherung vernetzter Peptide ermöglichen. Darüber hinaus führt die chemische Entfernung des Biotin-Tags zur Bildung einer funktionellen Gruppe, die bei der MS-Analyse einzigartige Reporterionen aus Leiker-vernetzten Peptiden erzeugt und so die Sicherheit ihrer Identifizierung unterstützt. Infolgedessen wurden 3.130 bzw. 893 Lysin-Verknüpfungen bei 5 % FDR aus E. coli und C. elegans identifiziert [124], was darauf hindeutet, dass die Vernetzungsanreicherung durch Affinitätsreinigung für die großtechnische XL-MS-Analyse von Vorteil ist.
5.2 In vivo proteomweite Studien
Im Vergleich zu in vitro-Studien können in vivo XL-MS-Experimente native Protein-Protein-Interaktionen besser erhalten, die dynamisch sind und stark von ihrer molekularen und zellulären Umgebung abhängen. Das Bruce-Labor und seine Mitarbeiter waren maßgeblich an der Anwendung von XL-MS-Strategien für in-culture-Studien beteiligt und haben ihre MS-spaltbaren, anreicherbaren PIR-Vernetzer erfolgreich für proteomweite Studien verschiedener Organismen, einschließlich Shewanella oneidensis [89], Escherichia coli [104], Pseudomonas aeruginosa [105] und Acinetobacter baumannii [106] sowie menschlicher Zellen [40, 77] eingesetzt. Jüngste Analysen von murinen Mitochondrien [107] führten zur Identifizierung von 2.427 vernetzten Peptidpaaren aus 327 mitochondrialen Proteinen mit einer FDR von 1,9 %. Neben PIR-Linkern wurde Azide-A-DSBSO [79] zur Kartierung von PPIs aus menschlichen Zellen durch Click-Chemie-basierte Konjugation und Biotin-basierte Anreicherung eingesetzt, während eine kürzlich durchgeführte Studie BAMG für die in vivo-Analyse von Bacillus subtilis [119] unter Verwendung einer Kombination aus Protein- und Peptid-Ebene-Trennung verwendete. Kollektiv haben diese Studien die Wirksamkeit der Kombination von Vernetzungsanreicherung und MS-Spaltbarkeit für in vivo-Studien demonstriert und eine solide Grundlage für zukünftige Bemühungen zur vollständigen Charakterisierung von PPI-Netzwerktopologien in lebenden Systemen geschaffen.
6. Quantitative XL-MS-Strategien und ihre Anwendungen
Die meisten Vernetzungsstudien zielten auf die Aufklärung statischer Proteinstrukturen ab. Proteinkomplexe sind jedoch von Natur aus dynamisch und existieren in einer Vielzahl von konformationellen und kompositorischen Zuständen – jeder mit seinen eigenen einzigartigen Eigenschaften, Merkmalen und Funktionen. Um die Interaktions- und Strukturdynamik von Proteinkomplexen zu analysieren, ist quantitative XL-MS (QXL-MS) erforderlich, um eine vergleichende Analyse verschiedener konformationeller Zustände von Proteinkomplexen zu ermöglichen, die unter verschiedenen physiologischen Bedingungen erhalten wurden. Änderungen der relativen Häufigkeiten einzelner Vernetzungen ermöglichen es uns, zustandsabhängige strukturelle Änderungen von Proteinen und Proteinkomplexen zu bewerten.
6.1 Markierung von Vernetzungsreagenzien
Ähnlich wie in der quantitativen Proteomik können stabile Isotope (d.h. 2H, 13C, 15N, 18O) in vernetzte Peptide eingebaut werden, um vergleichende Analysen durchzuführen. Die gängigste Strategie zur Markierung vernetzter Peptide für die Quantifizierung beinhaltet die Verwendung von isotopenkodierten Vernetzern (z.B. d0/d4-BS3 oder d0/d12-DSS). Vor ihren Anwendungen für QXL-MS wurden isotopenkodierte Vernetzer lange Zeit zur Erleichterung der Identifizierung nicht-spaltbarer vernetzter Peptide anhand ihrer einzigartigen isotopischen Peptidmuster verwendet [234]. Ihre Anwendung für QXL-MS wurde erstmals von den Gruppen von Rappsilber [235] und Robinson unter Verwendung von d0/d4-BS3 [236, 237] untersucht. Der allgemeine Workflow umfasst die separate Vernetzung der zu vergleichenden Proben, wobei eine mit dem leicht markierten Reagenz und die andere mit dem schwer markierten Reagenz reagiert (Abbildung 5A). Nach dem äquivalenten Mischen werden die Proteine verdaut und zur MS-Analyse und Datenbanksuche eingereicht. Die identifizierten vernetzten Peptide können auf MS1-Ebene quantifiziert werden, indem die relativen Häufigkeiten von leicht- und schwer markierten vernetzten Peptidpaaren gemessen werden. Durch den Vergleich der Vernetzungsprofile unbehandelter und dephosphorylierter Formen konnten Schmidt et al. die Auswirkungen der Phosphorylierung auf die Topologie der Spinat-Chloroplasten-ATPase unterscheiden und neue Einblicke in Phosphorylierung-vermittelte Regulationsmechanismen gewinnen [236, 237]. Während frühe QXL-MS-Studien eine manuelle Quantifizierung erforderten und daher in ihrer Anwendung begrenzt waren, wurden in den letzten Jahren erhebliche Fortschritte erzielt, um die Analyse von QXL-MS-Datensätzen zu ermöglichen. Software wie xTract [238] wurde speziell für die automatische Quantifizierung vernetzter Peptide entwickelt, während bestehende Tools wie MaxQuant [239], PinpointTM [240], pQuant [124] und mMass [241] für die Analyse von QXL-MS-Daten angepasst wurden (Tabelle 1). Die Automatisierung der QXL-MS-Analyse hat die Fähigkeit zur schnellen Analyse größerer Datensätze ermöglicht und das Spektrum quantitativer Studien von einzelnen Proteinen [240, 241] bis hin zu größeren Proteinkomplexen wie TRiC [238] und dem 19S-Proteasom-Lid [209] erweitert. Kürzlich demonstrierten Tan et al. die Machbarkeit der Extraktion von QXL-MS-Daten aus komplexen Gemischen unter Verwendung von d0/d6-Leiker-Vernetzer und pQuant [124].
 Workflows für proteomweite in vitro und in vivo QXL-MS-Experimente unter Verwendung MS-spaltbarer Vernetzer und MSn-basierter Akquisition.
Workflows für proteomweite in vitro und in vivo QXL-MS-Experimente unter Verwendung MS-spaltbarer Vernetzer und MSn-basierter Akquisition.
Obwohl die Mehrheit der Vernetzungsmarkierungs-basierten QXL-MS-Studien kommerziell erhältliche isotopenkodierte, nicht spaltbare Vernetzer (d.h. d0/d4-BS3 oder d0/d12-DSS) verwendet hat, haben wir versucht, quantitative Fähigkeiten mit MS-Spaltbarkeit zu integrieren, um eine vereinfachte, genaue Vernetzungsidentifizierung und -quantifizierung gleichzeitig zu ermöglichen. Daher entwickelten wir d0- und d10-markiertes DMDSSO, ein Paar isotopenkodierter, sulfoxidhaltiger MS-spaltbarer Vernetzer (Tabelle 2) [84]. Ähnlich wie DSSO werden d0/d10-DMDSSO-vernetzte Peptide in MS3 sequenziert, aber auf MS1-Ebene quantifiziert (Abbildung 5A). Unter Verwendung von Skyline und später unserer selbst entwickelten Software xl-Tools charakterisierten wir die Neddylierungs-abhängigen Konformationsänderungen, die mit der SCF-Ubiquitinligase-Aktivierung verbunden sind, und schlugen mechanistische Modelle für die Deaktivierung von SCFs durch bakterielle Effektorproteine mittels Nedd8-Deamidierung vor [96]. Darüber hinaus wurde kürzlich ein Satz isotopenmarkierter PIR-Vernetzer (d.h. d0/d8-BDP-NHP) für die quantitative Analyse in vivo vernetzter E. coli-Zellen eingesetzt [40], was zur Quantifizierung von 941 von 1213 identifizierten Peptidpaaren führte und die Machbarkeit von QXL-MS für die großtechnische Identifizierung und Quantifizierung weiter demonstrierte.
6.2 Metabolische Markierung
Zusätzlich zur Reagenzienmarkierung kann die Integration stabiler Isotope in vernetzte Peptide durch SILAC-Markierung erreicht werden [242]. Zufälligerweise ist Lysin nicht nur eine der am häufigsten verwendeten Aminosäuren für die SILAC-Markierung, sondern auch der beliebteste Zielrest für die Vernetzung. Ähnlich wie bei isotopenkodierten Reagenzienansätzen wird die SILAC-Quantifizierung auch durch den Vergleich der relativen Häufigkeiten von leicht- und schwer markierten vernetzten Peptidpaaren auf MS1-Ebene erreicht (Abbildung 5B). Im Vergleich dazu bieten SILAC-basierte Methoden nicht nur eine globale Markierung mit minimaler Varianz, sondern eliminieren auch die potenziellen Auswirkungen des Isotopeneinbaus auf Vernetzungsreaktionen und Peptidseparation. Isotopenkodierte Vernetzer werden am häufigsten mit Deuterium markiert, was ihre chromatographischen Profile im Vergleich zu ihren nicht markierten Gegenstücken verändert und die automatisierte Quantifizierung erschwert. Im Gegensatz dazu werden typischerweise 13C/12C- und 15N/14N-markierte Aminosäuren für die SILAC-Markierung verwendet, wodurch sowohl markierte als auch unmarkierte Vernetzungen chromatographisch koeluieren können. Leider erzeugt die metabolische Markierung auch Proben, die wesentlich komplexer sind als die Vernetzungsmarkierung, aufgrund des globalen Isotopeneinbaus (einschließlich nicht-vernetzter Peptide). Während es möglich ist, nur Lysine zu markieren, um vernetzte Peptide zu quantifizieren, ist eine SILAC-Markierung mit sowohl Lysin als auch Arginin für Vernetzer erforderlich, die auf andere Reste wie Asparaginsäure/Glutaminsäure abzielen, um die Quantifizierung aller tryptischen vernetzten Peptide zu gewährleisten. Obwohl SILAC-basierte Methoden im Allgemeinen auf Zellkultursysteme beschränkt sind, kann die Idee, markierte Zellkulturen als interne Referenzen zum Vergleich nicht-markierter Proben in proteomischen Studien [243] potenziell übernommen und mit QXL-MS für erweiterte Anwendungen integriert werden. Während SILAC-basierte Methoden seit mehr als einem Jahrzehnt in der quantitativen Proteomik weit verbreitet sind [242, 244], wurden sie erst kürzlich mit der XL-MS-Analyse integriert [40, 108, 139, 150]. SILAC-basierte QXL-MS-Ansätze wurden zur Quantifizierung in vivo von Interaktionen und struktureller Dynamik von Hsp90 und Proteomen als Reaktion auf Medikamentenbehandlungen angewendet [108, 150]. Diese Anwendungen haben eine solide Grundlage für zukünftige QXL-MS-Experimente unter Verwendung metabolischer Markierung geschaffen.
6.3 Isobare Markierung für multiplexierte Quantifizierung
Obwohl sowohl Vernetzungs- als auch SILAC-basierte QXL-MS-Strategien erfolgreiche quantitative Studien ermöglicht haben, sind diese Workflows typischerweise auf binäre (paarweise) Vergleiche beschränkt, aufgrund unzureichender differenzierender Isotopenmarkierungen, erhöhter spektraler Komplexität und verringerter Nachweisbarkeit von gering abundanten vernetzten Peptiden. Inspiriert durch den enormen Erfolg der multiplexierten Quantifizierung in proteomischen Studien unter Verwendung isobarer Markierungsreagenzien (z.B. iTRAQ [245] und TMT-Reagenzien [246, 247]), haben wir kürzlich versucht, die multiplexierte Fähigkeit auf die QXL-MS-Analyse auszudehnen [94]. Isobare Markierungs-basierte quantitative Methoden ermöglichen die parallele Analyse von bis zu 11 Experimenten mit kommerziell erhältlichen Reagenzien (d.h. TMT 11-plex), wodurch der Durchsatz und die Quantifizierungsgenauigkeit ohne Erhöhung der Probenkomplexität erheblich gesteigert werden. Im Gegensatz zu isotopenmarkierungsbasierten Methoden wird die Quantifizierung durch die Detektion einzigartiger Reporterionen erreicht, die aus der Fragmentierung isobarer Markierungen während der HCD auf MS2-Ebene resultieren. Da alle Peptide durch dieselben isobaren Markierungen modifiziert sind, kann während der MS2-Ebene-Quantifizierung eine Peptidinterferenz auftreten, die zu einer Unterschätzung quantitativer Änderungen zwischen den verglichenen Proben führt [248]. Diese Verzerrung kann durch die Quantifizierung in MS3 [248] effektiv eliminiert werden, wo die Intensitäten der Reporterionen durch die SPS-Akquisition (Synchronous Precursor Selection) erheblich verstärkt werden können, um die Quantifizierungsgenauigkeit zu erhöhen [249]. Um einen robusten Workflow zu etablieren, der die vergleichende Analyse mehrerer vernetzter Proben gleichzeitig ermöglicht, haben wir eine multiplexierte QXL-MS-Strategie entwickelt, nämlich QMIX (Quantitation of Multiplexed, Isobaric-labeled cross (X)-linked peptides), indem wir MS-spaltbare Vernetzer mit isobaren Markierungsreagenzien integrieren (Abbildung 5C) [94]. In einer Proof-of-Concept-Studie kombinierten wir DSSO-basierte XL-MS mit TMT-Reagenzien und zeigten, dass die Identifizierung und Quantifizierung isobar markierter, vernetzter Peptide gleichzeitig in MS3 durchgeführt werden kann [94]. Die etablierte QMIX-Strategie wird die hochdurchsatzfähige vergleichende Analyse erleichtern, um Interaktions- und Strukturdynamiken von Proteinkomplexen und Proteomen zu bestimmen. Es ist zu beachten, dass QMIX eine allgemeine Strategie ist, die mit allen Vernetzungsreagenzien kompatibel ist, unabhängig von ihren restpezifischen Chemien oder chemischen Funktionalitäten. Obwohl die isobare Markierung theoretisch auf konventionelle nicht-spaltbare Vernetzer angewendet werden kann, ist die Kopplung dieser Multiplexierungsstrategie mit MS-spaltbaren Vernetzungsreagenzien aufgrund der Kompatibilität von MSn-basierten Akquisitionen und der daraus resultierenden Vereinfachung der Identifizierung vernetzter Peptide optimal [94].
7. Zukunftsperspektiven und Schlussfolgerungen
Fünf entscheidende Schritte sind normalerweise bei XL-MS-Studien für Interaktomik und Strukturbiologie involviert: 1) die Wahl der Vernetzer; 2) die Probenvorbereitung; 3) die MS-Analyse; 4) die Datenverarbeitung; 5) die Interaktionskartierung und strukturelle Charakterisierung. Die Weiterentwicklung der XL-MS-Analyse beruhte auf Verbesserungen und Innovationen in jedem dieser Aspekte, was die heutige XL-MS zur Methode der Wahl für die Definition von PPI-Landschaften auf Systemebene und die Aufklärung der Architekturen großer Proteinassemblierungen gemacht hat. Mit dem Aufkommen robuster Vernetzungsreagenzien, ultra-leistungsfähiger Massenspektrometer und leistungsstarker Bioinformatik-Tools ist die XL-MS-Analyse für die Forschungsgemeinschaft viel zugänglicher und machbarer geworden als zuvor. Während das XL-MS-Feld in das Rampenlicht der Proteomforschung tritt, sind immer noch neue Entwicklungen in allen fünf Schlüsselbereichen erforderlich, um das Feld weiter zu reifen und XL-MS-Experimente für die Routineanalyse mit Empfindlichkeit, Genauigkeit, Zuverlässigkeit, Tiefe, Geschwindigkeit und Durchsatz zu ermöglichen. Während konventionelle nicht-spaltbare Vernetzer mit Hilfe neuer Datenbank-Suchwerkzeuge weiterhin eine Rolle in XL-MS-Studien spielen werden, erwarten wir, dass MS-spaltbare Vernetzer weiterhin dazu beitragen werden, vereinfachte und robuste XL-MS-Workflows für die großtechnische PPI-Profilierung in vitro und in vivo voranzutreiben. Die kommerzielle Verfügbarkeit von MS-spaltbaren Vernetzern wie DSSO, DSBU und CDI wird sicherlich verschiedene Anwendungen von MS-spaltbaren Vernetzern in naher Zukunft erleichtern. Darüber hinaus wird die Erforschung neuer Vernetzungschemien die XL-MS-Toolbox um zusätzliche Fähigkeiten erweitern, um bestehende Reagenzien zu ergänzen. Dies ist wichtig, da schnellere Kinetiken in Vernetzungsreaktionen eine bessere Erfassung transienter/dynamischer Interaktionen in Zellen ermöglichen und neue restsäurezielende Chemien eine breitere Abdeckung von Interaktionsflächen erlauben werden. Während die jüngste Bewertung nicht-spezifischer Vernetzer mit Einzelproteinen vielversprechende Ergebnisse bei der Gewinnung einer breiten Palette von Abstandsrestriktionen gezeigt hat [137], müssen die Machbarkeit und das Potenzial dieser Reagenzien in der XL-MS-Analyse komplexer Systeme sorgfältig geprüft werden. Obwohl Peptid-Reinigungsstrategien, die auf anreicherbaren Vernetzern und Trennungstechniken basieren, entwickelt wurden, sind weiterhin analytische Verbesserungen in Empfindlichkeit, Spezifität und Selektivität erforderlich, um die MS-Detektierbarkeit und Identifizierung von gering abundanten vernetzten Peptiden in komplexen Gemischen weiter zu verbessern. Während der Biotin-Tag ausschließlich für die Reinigung vernetzter Peptide verwendet wurde, deutet ein kürzlich veröffentlichter Bericht [130] darauf hin, dass auch andere Affinitäts-Tags (z.B. CH-Tag) für spezifische XL-MS-Anwendungen nützlich sein könnten. Dies impliziert, dass Strategien zur Protein-Affinitätsreinigung für die Peptidanreicherung übernommen werden könnten. Die Zugänglichkeit mehrerer Peptidfragmentierungstechniken (z.B. CID, HCD, ETD, EThCD, UVPD) bietet mehr Flexibilität in der MS-Analyse und hat es ermöglicht, bestehende Vernetzer (z.B. DSSO) neu zu untersuchen und so neue komplementäre Workflows (Abbildungen 2A und 2B) zur Gewinnung umfassenderer Vernetzungsdaten zu etablieren. Die Kombination verschiedener Fragmentierungstechniken würde nicht nur das Potenzial bekannter Vernetzer maximieren, sondern auch Anleitungen für zukünftige Designs neuer Vernetzungsreagenzien geben.
Abgesehen von der statischen Strukturanalyse stellt die QXL-MS die nächste Generation der XL-MS dar, um die Charakterisierung dynamischer Proteome unter verschiedenen physiologischen Bedingungen in verschiedenen Organismen zu ermöglichen. Es wurde gezeigt, dass Methoden der quantitativen Proteomik direkt auf die QXL-MS-Analyse übertragen werden können, was deren Entwicklung und Anwendungen erheblich erleichtert. Zusätzlich zur paarweisen Quantifizierung wurden kürzlich multiplexierte QXL-MS-Plattformen auf der Grundlage isobarer Markierung etabliert [94]. Darüber hinaus wurde die Machbarkeit der PRM (Parallel Reaction Monitoring)-basierten gezielten Quantifizierung auch für XL-MS-Studien illustriert [40, 139]. Daher gehen wir davon aus, dass die Kombination experimenteller Entwicklungen mit leistungsstarker multiplexierter relativer und gezielter Quantifizierung quantitative Bewertungen der PPI-Dynamik auf Proteinkomplex- und Proteom-Ebene mit Geschwindigkeit und Genauigkeit ermöglichen wird. Es bestehen jedoch Herausforderungen hinsichtlich der automatisierten Datenverarbeitung, um eine schnelle und genaue Identifizierung und Quantifizierung gleichzeitig mit mehrschichtigen Daten, die in verschiedenen MS-Stufen erfasst wurden, zu ermöglichen. Darüber hinaus muss sorgfältig geprüft werden, wie relative Abundanzänderungen in Vernetzungen korrekt interpretiert werden, um Proteininteraktionen und Konformationsänderungen abzuleiten, da die Vernetzbarkeit von Resten von vielen Faktoren abhängt, einschließlich ihrer Nähe, chemischen Umgebungen und Lösungsmittelzugänglichkeit. Dieser Prozess kann jedoch durch Strukturmodellierungswerkzeuge erleichtert werden, um die Art der quantitativen Änderungen besser zu analysieren. Andererseits können die gemessenen quantitativen Änderungen von Vernetzungen als eine weitere Art von Beschränkungen dienen, um aus Abstandsrestriktionen abgeleitete Strukturmodelle zu verfeinern. Es wird deutlich, dass eine kontinuierliche Weiterentwicklung der XL-MS-Software dringend erwartet wird, um die Vernetzungsidentifizierung, -quantifizierung und -validierung sowie die Visualisierung, Dateninterpretation und Modellierung zu beschleunigen.
Während große Anstrengungen in in vitro-Studien unternommen werden, sind in vivo-Anwendungen das ultimative Ziel für XL-MS-Technologien. Zahlreiche Beweise haben gezeigt, dass abweichende Protein-Protein-Interaktionen zu menschlichen Krankheiten und Krebs führen können und Proteininteraktionsflächen eine neue Klasse attraktiver Ziele für die Arzneimittelentwicklung darstellen [250–253]. Daher wird die Fähigkeit, authentische PPI-Netzwerkkarten zu erhalten und deren zeitliche und räumliche Dynamik aus lebenden Systemen zu definieren, nicht nur molekulare Details aufdecken, die der Funktion und Regulation makromolekularer Maschinen zugrunde liegen, sondern auch ein einzigartiges Mittel zur Unterstützung der Entwicklung interaktionsabhängiger Therapeutika bieten. Mit kontinuierlichen Durchbrüchen auf diesem Gebiet erwarten wir, dass XL-MS weitere bedeutende Beiträge zur Strukturbiologie, aber vor allem zur fortgeschrittenen biomedizinischen Forschung leisten wird.