
Die ständige Weiterentwicklung von Viren stellt eine anhaltende Herausforderung für die globale Gesundheitskontrolle und Impfstoffentwicklung dar. Insbesondere das SARS-CoV-2-Virus hat mit seinen schnellen Mutationen die wissenschaftliche Gemeinschaft weltweit beschäftigt. Unser Team hat eine eingehende spatiotemporale Analyse von Intra-Host-Single-Nukleotid-Varianten (iSNVs) in über 400 klinischen Proben von 170 infizierten Personen durchgeführt. Diese Untersuchung enthüllt eine signifikante Zunahme der genetischen Diversität im Laufe der Zeit nach dem Auftreten von Symptomen. Faszinierenderweise sind nicht-synonyme Mutationen in der Population der iSNVs überrepräsentiert, während sie auf der Ebene von Single-Nukleotid-Polymorphismen (SNPs) unterrepräsentiert sind. Dies deutet auf einen zweistufigen Prozess der “Step Fitness”-Selektion hin: Zuerst wird eine große Anzahl nicht-synonymer Substitutionen im Wirt generiert (positive Selektion), und anschließend werden diese Substitutionen in der Gesamtpopulation eher nicht fixiert (negative Selektion). Die dynamischen Veränderungen von iSNVs, die sich in Unterpopulationen mit unterschiedlichem Geschlecht, Alter, Krankheitsverlauf und viraler Ausscheidungsdauer zeigen, weisen auf differenzierte “Step Fitness”-Selektionsprozesse hin. Unsere Studie unterstreicht die entscheidende Rolle von iSNVs als mutativen Pool, der die schnelle globale Evolution des Virus maßgeblich prägt.
Die rasante Ausbreitung des SARS-CoV-2-Virus hat trotz weltweiter Präventions- und Kontrollmaßnahmen weltweit für Besorgnis gesorgt. Als einzelsträngiges RNA-Virus weist SARS-CoV-2 eine hohe Mutationsrate auf, und unerwartete Mutationen können das Virus verändern und seine Kontrolle erschweren, was zu einer verminderten Impfstoffwirksamkeit führen kann. Tausende von Mutationen wurden bereits identifiziert, die zur Entstehung neuer viraler Linien wie Alpha, Delta und Omikron geführt haben. Die kontinuierliche Überwachung dieser Mutationen in den verschiedenen Viruslinien ist daher von globaler Priorität.
Die Entstehung von Intra-Host-Varianten (iSNVs)
Mutationsereignisse entstehen zufällig in einem kleinen Bruchteil von Viren innerhalb eines einzelnen infizierten Wirts und generieren so Intra-Host-Single-Nukleotid-Varianten (iSNVs). Einige dieser iSNVs können später im Wirt fixiert und zwischen Populationen übertragen werden oder in nicht-fixierter Form weitergegeben werden, was schließlich zur Entstehung genetisch diverser Populationen führt. Während der Virusausbreitung werden iSNVs sowohl durch stochastische Prozesse (z.B. genetische Drift) als auch durch deterministische Prozesse (z.B. “Step Fitness”-Selektion) fixiert. Hochdurchsatz-Sequenzierungstechniken ermöglichen es uns, diese Prozesse zu untersuchen und die genetische Diversität auf Populationsebene sowie innerhalb des einzelnen Wirts zu quantifizieren. Frühere Studien auf Populationsebene, die klinische, molekulare und immunologische Daten von SNPs nutzten, haben bereits wesentliche Einblicke in die Epidemiologie, Krankheitsübertragung und Pathogenese geliefert.
iSNVs: Ein tieferer Einblick in die virale Evolution
iSNVs, als umfangreicherer genetischer Mutationspool im Vergleich zu SNPs, liefern zusätzliche Informationen, die die Diversität und Dynamik der Virusentwicklung in einzelnen Wirten definieren. Die Analyse von iSNVs ergänzt konventionelle SNP-Studien auf Populationsebene und ermöglicht ein umfassenderes Verständnis der Virusentwicklung. Dies ist entscheidend für klinisch relevante Vorhersagen der Virusentwicklung im Zusammenhang mit Infektionen, Pandemien und Immun-Evasion. Bisherige Studien, die sich mit den genetischen Merkmalen von iSNVs bei COVID-19-Patienten befassten, sind jedoch begrenzt.
In unserer Studie haben wir die genetische Diversität von SARS-CoV-2 und die Virusentwicklung in einzelnen Wirten quantitativ bewertet, indem wir tiefe virale Genomsequenzierung und empirische Analyse-Pipelines einsetzten. Eine spatiotemporale Analyse der Genomdaten zeigte, dass die Variation innerhalb des Wirts nicht zufällig über das Genom verteilt war und dass solche Variationen die genetische Diversität von SARS-CoV-2 erhöhten, was auf eine Rolle der Selektion hindeutet. Nicht-synonyme Mutationen waren in iSNVs überrepräsentiert (Mutationsallelfrequenz [MuAF], >5% und <95%), aber in der Menge der SNPs (MuAF, >95%) unterrepräsentiert. Dies deutet auf einen zweistufigen “Step Fitness”-Selektionsprozess hin. Wir untersuchten auch die Auswirkungen von iSNVs (nicht-fixierte Mutationen) auf klinische Charakteristika und die Bindungsaffinität des Spike (S)-Proteins, was den beobachteten gerichteten Selektionsprozess erklären könnte. Unsere Ergebnisse legen nahe, dass es wichtig ist, gleichzeitig die Dynamik innerhalb und zwischen Wirten bei aufkommenden Viren zu untersuchen, um deren Evolutionsgeschichte zu verstehen und gezielte Präventions- und Kontrollmaßnahmen zu entwickeln.
Identifizierung von Intra-Host-Varianten in SARS-CoV-2-Genomen
Wir sammelten 537 Proben (183 Pharyngeal-, 241 Sputum- und 113 Fäkalproben) von 204 betroffenen Personen, was 34,4% der Gesamtfälle in Peking vor dem 30. April 2020 abdeckt. Mittels gezielter Capture-Sequenzierung des viralen Genoms erhielten wir durchschnittlich 8,59 G Basenpaare pro Probe. Davon wurden 11,90% auf das Referenzgenom von SARS-CoV-2 Wuhan-Hu-1 abgebildet. Proben mit geringer viraler Genomabdeckung wurden entfernt, und schließlich wurden 402 Proben von 170 betroffenen Personen für die weitere Analyse ausgewählt. Die Sequenziertiefe betrug 28.720x über das gesamte Genom. Von diesen Personen hatten 81 mindestens zwei verschiedene Probentypen oder Entnahmezeitpunkte.
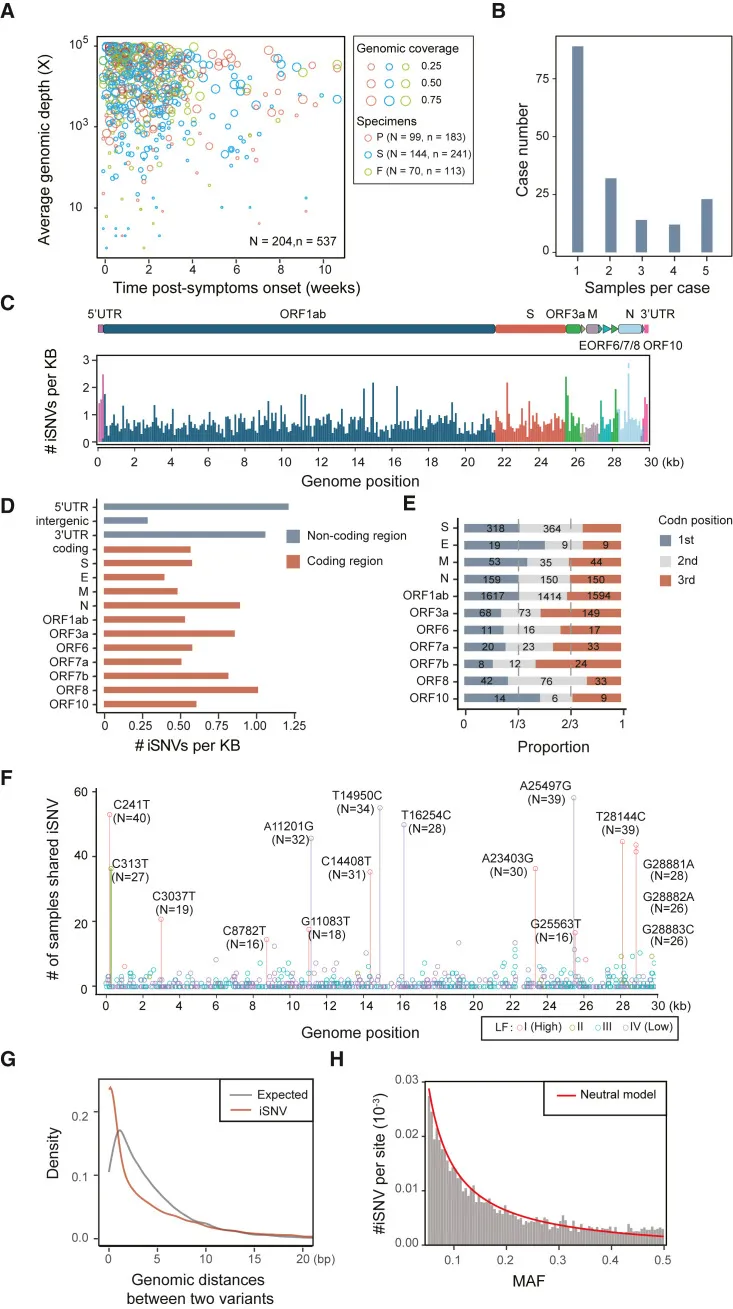 Genomsequenzierung und iSNV-Loci
Genomsequenzierung und iSNV-Loci
Für jede Probe führten wir eine hochauflösende Untersuchung von SARS-CoV-2-Genomstellen auf iSNVs durch (mindestens 100x Tiefe), die mit einem strengen Schwellenwert (≥5%) gefiltert wurden, um echte iSNVs von Sequenzierungsfehlern zu unterscheiden. Wir validierten die Probenverarbeitungspipeline mit technischen Replikaten in 62 Proben mit unabhängiger Bibliotheksvorbereitung. Unter dem 5%-Schwellenwert für MuAF identifizierten wir 450 reproduzierbare iSNVs von 498 iSNVs in den ersten Experimenten. Die iSNVs, die diesen strengen Schwellenwert erfüllten, waren über das gesamte Genom verteilt, und die Anzahl der iSNVs pro Probe wurde weder durch die Genomabdeckung noch durch die Sequenziertiefe beeinflusst. Insgesamt identifizierten wir 7.037 iSNVs in 374 Proben mit einer mittleren Dichte von 0,53 iSNVs/kb, was mit der Anzahl der zuvor für SARS-CoV-2 berichteten iSNVs und anderen Viren wie dem Ebola-Virus vergleichbar ist. Etwa 93% der Proben (374 von 402) enthielten mindestens ein iSNV im Vergleich zum Referenzgenom.
Ungleichmäßige Verteilung von Intra-Host-Varianten
Wir untersuchten die Lokalisation der iSNVs entlang des SARS-CoV-2-Genoms und stellten eine insgesamt relativ geringe iSNV-Dichte (0,58 iSNVs/kb) fest, die mit früheren Berichten vergleichbar ist. Höhere iSNV-Dichten wurden im 5′-UTR (1,23 iSNVs/kb) und 3′-UTR (1,07 iSNVs/kb) beobachtet. Wir fanden 6.790 (96,49%) iSNVs in kodierenden Regionen, die 97,85% des gesamten Genoms ausmachen. Die meisten iSNVs (4.625, 68,11%) wurden in offenen Leserahmen (ORF) 1ab identifiziert, gefolgt vom S-Gen (903 iSNVs) und dem N-Gen (459 iSNVs). Nach der Normierung der iSNVs auf die Genlänge wurde jedoch die höchste Häufigkeit von iSNVs in ORF8 (1,02 iSNVs/kb) gefunden, gefolgt vom N-Gen (0,906 iSNVs/kb). Diese Ergebnisse stimmten mit einer früheren Studie zu SARS-CoV-2 auf SNP-Ebene überein. Daher könnte die Ungleichgewichtsverteilung von Mutationen zwischen den Genen auf iSNV-Ebene auftreten und im Fixierungsprozess aufrechterhalten werden. Darüber hinaus zeigte die Analyse der Codon-Positionen 2.329, 2.178 und 2.283 iSNVs an der ersten, zweiten bzw. dritten Codon-Position. Ein Fisher-Exaktest ergab, dass ORF10 und das E-Gen eine signifikant größere Anzahl von iSNVs an der ersten Codon-Position aufwiesen im Vergleich zu den anderen Codon-Positionen in koordinierten Genen.
Wir untersuchten als Nächstes die Verteilung der iSNVs unter den Individuen. Von den 4.690 iSNV-Stellen wurden 81,02% nur bei einem einzigen Individuum beobachtet, und 18,98% wurden bei mindestens zwei Individuen gefunden, was mit einem früheren Bericht vergleichbar ist. Es gab 16 hochgradig rezidivierende iSNVs, die bei mindestens 15 Individuen gemeinsam auftraten. Unter diesen überschnitten sich 12 Stellen mit hochfrequenten SNP-Stellen (hfSNPs), die zuvor in der globalen SARS-CoV-2-Genomdatenbank 2019nCoVR basierend auf mehr als 5% der Individuen definiert worden waren. Dieses Phänomen wurde auch in einem früheren Bericht beschrieben und könnte auf konvergente positive Selektion oder mutative Hotspots zurückzuführen sein. Wir konstruierten einen einfachen Rahmen zur Berechnung der Verteilung der Genomdistanz zwischen Allelen von iSNVs, und die angepasste Dichtelinie zeigte eine signifikante Abweichung von zufällig generierten Mutationen (Kolmogorov-Smirnov-Test, p < 0,001), was auf die Nicht-Stochastizität der iSNV-Verteilung hindeutet. Darüber hinaus prognostizierten wir basierend auf einer Modifikation des stochastischen Geburts-Tod-Modells Allelfrequenzen, um zu beschreiben, wie Mutationen mit der Expansion von Zellen akkumulieren. Obwohl die Verteilung ähnlich dem neutralen Modell war, waren die iSNV-Anzahlen in den hochfrequenten Allelen größer als die erwarteten Werte im neutralen Evolutionsmodell, was auf eine potenzielle positive Selektion von iSNV-Stellen hindeutet.
Genetische Diversität steigt mit fortschreitender Krankheit
Um dynamische Veränderungen der viralen iSNVs bei COVID-19-Patienten aufzudecken, führten wir eine spatiotemporale Analyse von iSNVs über den Epidemiezeitraum und den Krankheitsverlauf anhand verschiedener Proben durch. Zunächst beobachteten wir eine stetige Zunahme der Anzahl von iSNVs im Laufe der Zeit während der Epidemie (geschätzter Wert von 0,15 bis 0,83 iSNVs/kb innerhalb von 97 Tagen). Ähnliche Zunahmen der iSNV-Anzahlen wurden in anderen viralen Genomen beobachtet, die wir untersuchten und die akute und chronische Infektionen verursachen (Gelbfiebervirus, Zika-Virus, menschliches Immundefizienz-Virus, Hepatitis-C-Virus). Darüber hinaus akkumulierten sich iSNVs auch während der Infektion im einzelnen Wirt, von 0,52 iSNVs/kb am Tag 1 bis 0,85 iSNVs/kb 30 Tage nach Symptombeginn. Die Akkumulation war sogar nach Normierung auf virale RNA-Zyklusschwellenwerte (Ct-Werte) nachweisbar.
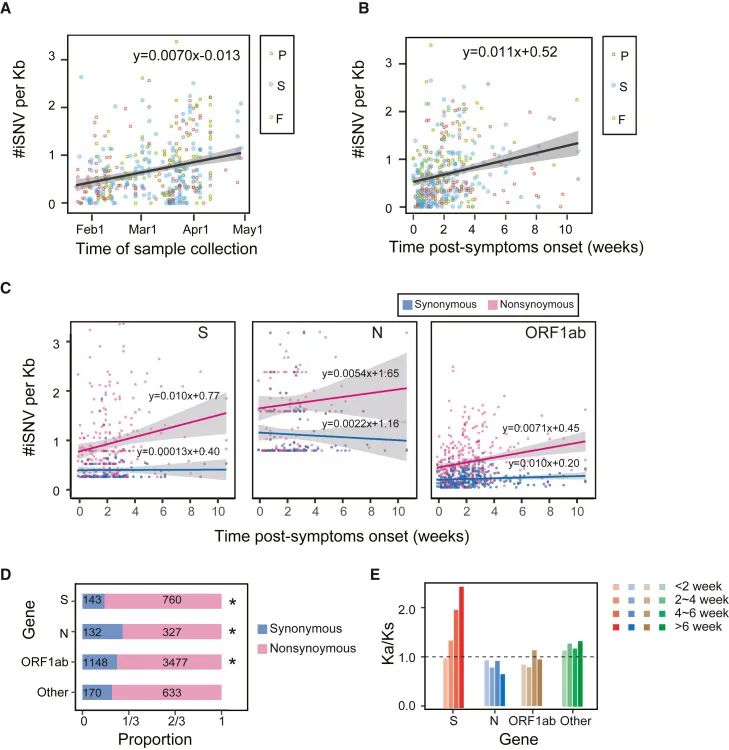 Spatiotemporale Analyse von iSNVs
Spatiotemporale Analyse von iSNVs
Die räumliche Analyse der drei Probentypen, die verschiedene Körperstellen/Orte repräsentieren, zeigte jedoch eine erhöhte genetische Diversität in allen drei Proben entlang des Krankheitsverlaufs. Wir identifizierten mehr iSNVs am Anfangstag und eine geringere Akkumulationsrate während des Krankheitsverlaufs in Fäkalproben im Vergleich zu Pharyngealabstrichen und Sputumproben (p = 0,006, ANOVA).
Positiver Selektionsprozess für iSNVs
Um zu prüfen, ob die beobachtete Akkumulation genetischer Diversität durch “Step Fitness”-Selektion verursacht wurde, untersuchten wir die dynamische Veränderung von nicht-synonymen und synonymen Mutationen mit dem Krankheitsverlauf in den S-, N-, ORF1ab- und anderen Genen. Im Vergleich zu neutralen synonymen Mutationen war die Akkumulation von Mutationen in nicht-synonymen Regionen in allen Genen schneller, und das S-Gen zeigte die höchste Akkumulationsrate. Entlang des Genoms stellten wir fest, dass 5.197 iSNVs nicht-synonyme Mutationen waren, während nur 1.593 iSNVs synonyme Mutationen waren. Das Verhältnis von nicht-synonymen zu synonymen Varianten bei allen Individuen betrug 3,26 (mittleres Verhältnis von 3,16 pro Individuum). Das Verhältnis von nicht-synonymen zu synonymen iSNVs divergiert zwischen den Genen; das Verhältnis im S-Gen (Verhältnis = 5,31) war signifikant höher als in den anderen Teilen des viralen Genoms (p < 0,001, Fisher-Exaktest). Die Durchschnittswerte der Minor-Allelfrequenzen von nicht-synonymen und synonymen iSNVs betrugen 0,189 bzw. 0,195. Mit einem einfachen Substitutionsmodell verwendeten wir das Verhältnis von Ka/Ks, um zu messen, ob die Gene im SARS-CoV-2-Genom unter Selektionsdruck standen. Das Ka/Ks-Verhältnis im S-Gen stieg von 1,01 auf 2,46 mit fortschreitender Krankheit, was darauf hindeutet, dass mit fortschreitender Krankheit eine positive Selektion stattfand, zumindest für das S-Gen.
Zusätzlich nutzten wir zwei Datensätze aus der Immune Epitope Database (IEDB) (experimentell bestätigte und vorhergesagte Epitopregionen), um den Anteil nicht-synonymer/synonymer Mutationen innerhalb und außerhalb von Epitopregionen zu bewerten. Der Anteil nicht-synonymer Mutationen in den Epitopregionen war signifikant höher als erwartet (27,57 % gegenüber 25,74 %, p = 0,016, Fisher-Exaktest). Entsprechend waren nicht-synonyme Mutationen außerhalb von Epitopregionen signifikant seltener. Eine weitere Korrelationsanalyse zwischen dem Anteil nicht-synonymer Stellen in vorhergesagten Epitopregionen und dem Zeitpunkt des Symptombeginns ergab, dass die Anzahl nicht-synonymer Stellen im S-Gen mit fortschreitender Krankheit zunahm.
Schließlich untersuchten wir die Dynamik der intra-host-Evolution in 268 Proben von 61 longitudinal beprobten Individuen. Die Zeitintervalle zwischen der Entnahme der ersten und letzten Probe betrugen mehr als 5 Tage. Keiner der Individuen wurde einer Antikörper- oder Immunsuppressivbehandlung unterzogen. Obwohl die Mutationsmuster über die Zeit individuell variierten, zeigten die meisten Individuen (45 von 61) eine erhöhte Mutationsvielfalt. Wir schätzten die Akkumulationsrate für jedes Individuum mithilfe eines linearen Modells der iSNV-Anzahl und der Zeit nach Symptombeginn. Im Einklang mit den schnell akkumulierten iSNVs an nicht-synonymen Stellen zeigten 84,44 % der Individuen (38 von 45) höhere Akkumulationsraten an nicht-synonymen Stellen als an synonymen Stellen. Eine kleine Anzahl stabiler iSNV-Stellen (81 von 3.629) trat wiederholt über die Zeitpunkte hinweg auf. Darüber hinaus identifizierten wir 255 rezidivierende iSNVs zu verschiedenen Zeitpunkten. Unter diesen zeigten 143 iSNVs erhöhte Allelfrequenzen und die verbleibenden 112 iSNVs zeigten verringerte Allelfrequenzen zu späteren Zeitpunkten, was stärkere Hinweise auf eine potenzielle positive Selektion innerhalb des Wirts liefert.
RNA-Editing in Regionen erhöhter genetischer Diversität
RNA-Editing-Enzyme können einzelsträngige RNA- und DNA-Moleküle mutagenisieren und einen Abwehrmechanismus gegen Viren darstellen. Zwei Familien von RNA-Editing-Enzymen haben sich als beitragend zum mutativen Spektrum von SARS-CoV-2 erwiesen. Die Apolipoprotein B mRNA-Editing-katalytische Polypeptid-ähnliche Deaminase (APOBEC) deaminisiert Cytosine zu Uracil (C zu U, einschließlich C zu U und G zu A), während die Adenosin-Deaminase (RNA-spezifisch) (ADAR) Adenosine zu Inosinen deaminisiert (A zu I, einschließlich A zu G und U zu C).
Wir maßen alle Mutationstypen für alle iSNVs, und die Top-Fünf-iSNV-Mutationstypen wurden wie folgt eingestuft (am häufigsten bis am wenigsten häufig): U zu C, C zu U, G zu U, A zu G und G zu A. Vier davon könnten durch APOBEC und ADAR eingeführt worden sein. Im Gegensatz zum A-zu-I-RNA-Editing-Signal im menschlichen Transkriptom beobachteten wir keine offensichtliche Verringerung von G-Basen an Position −1. Um die dynamische Veränderung der RNA-Editing-Niveaus im Einklang mit dem Krankheitsverlauf zu bewerten, berechneten wir die Korrelationen zwischen den Minor-Allelfrequenzen von iSNVs und der Zeit nach Symptombeginn. Die Minor-Allelfrequenzen von C-zu-U- und G-zu-A-Mutationen, die auf APOBEC-vermitteltes RNA-Editing zurückzuführen sind, nahmen in vivo leicht zu. Im Gegensatz dazu nahmen die Frequenzen anderer Mutationstypen, einschließlich ADAR-vermitteltem RNA-Editing in vivo, mit der Infektionsdauer nicht zu. Diese Identifizierung stimmte mit früheren Studien überein, die feststellten, dass Coronavirus-Infektionen APOBEC-Aktivität, aber nicht ADAR-Aktivität induzieren. Wir verglichen als Nächstes die Mutationsarten und ihre Akkumulationsraten für nicht-synonyme und synonyme Mutationen. Alle vier durch APOBECs/ADARs vermittelten Substitutionen waren bei nicht-synonymen Mutationen häufiger. Darüber hinaus akkumulierten sich synonymer Mutationen schneller im Vergleich zu nicht-synonymen Mutationen. Diese Ergebnisse legen nahe, dass RNA-Editing, vermittelt durch APOBECs, auch von SARS-CoV-2-Infektionen beeinflusst wurde, insbesondere die Raten von C-zu-U- und G-zu-A-Mutationen.
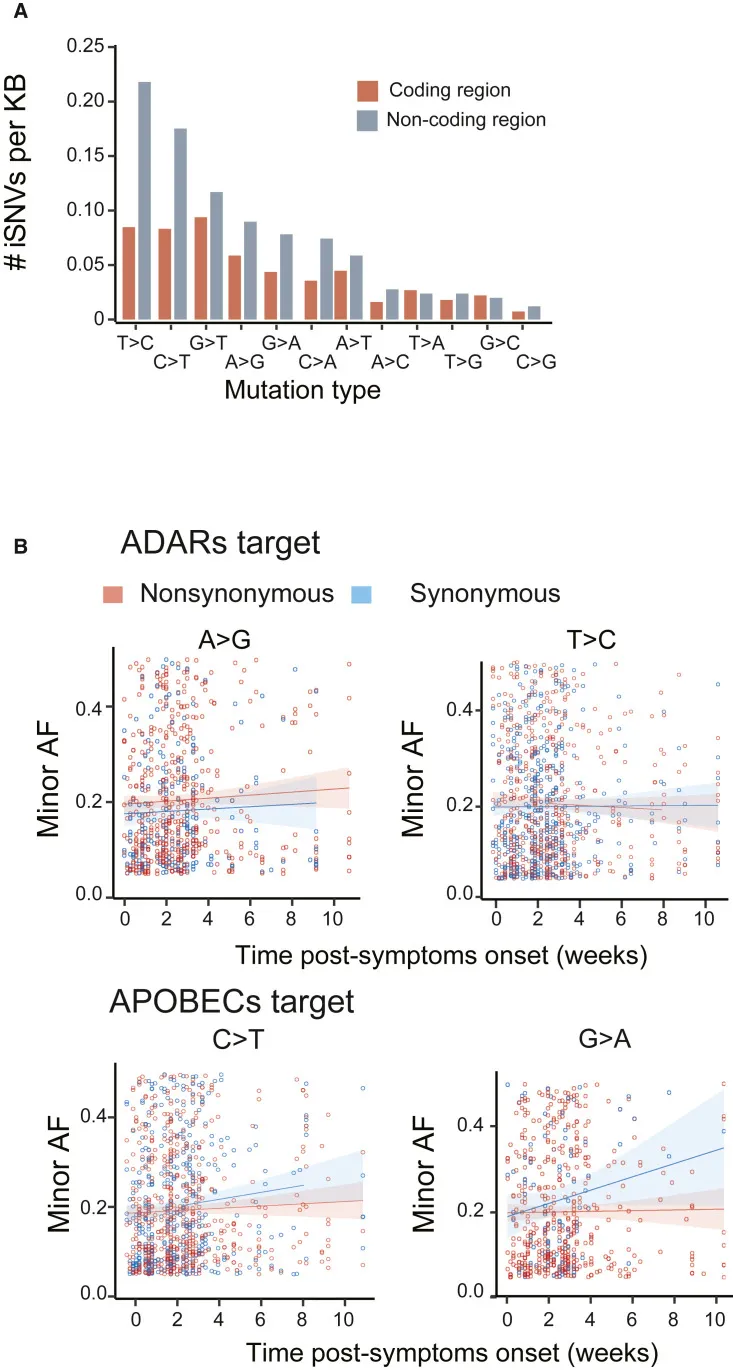 iSNV-Verteilungen verschiedener Mutationstypen
iSNV-Verteilungen verschiedener Mutationstypen
Einfluss möglicher Wirtseffekte auf die genetische Diversität
Eine verschlimmerte Entzündungsreaktion wurde bei schweren und kritischen Patienten beobachtet, und unterschiedliche Immunitätsprofile wurden bei verschiedenen Geschlechts- und Altersgruppen berichtet. Daher bewerteten wir den Einfluss von Wirtseffekten auf virale Mutationen und maßen dynamische Veränderungen der Anzahl von iSNVs innerhalb von Patientengruppen basierend auf Geschlecht, Alter, Krankheitschwere und viraler Ausscheidungsdauer. Jede Probe wurde anhand des Symptombeginns neu kalibriert. Eine erhöhte genetische Diversität von iSNVs wurde in allen Gruppen beobachtet, was darauf hindeutet, dass die Akkumulation von iSNVs in allen Populationen auftrat und nicht in einer spezifischen Population. Wir beobachteten auch unterschiedliche Steigungen und Anfangswerte im “Step Fitness”-Linearmodell zwischen der iSNV-Anzahl und der Zeit nach Symptombeginn in diesen Gruppen. Eine höhere Akkumulationsrate wurde bei mittelalten Individuen (15–65 Jahre) im Vergleich zu älteren Personen (p = 0,037, ANOVA) beobachtet, insbesondere bei denen, deren virale Ausscheidungsdauer mehr als 6 Wochen betrug (p = 0,041, ANOVA). Wir beobachteten auch eine signifikante Zunahme nicht-synonymer Stellen bei mittelalten im Vergleich zu älteren Individuen (p = 0,035, ANOVA-Test), während die Rate der synonymen Akkumulation zwischen verschiedenen Altersgruppen nicht unterschiedlich war. Die unterschiedliche iSNV-Akkumulationsrate deutet auf das Vorhandensein eines anderen “Step Fitness”-Selektionsprozesses während der anfänglichen Infektionsphase und der nachfolgenden Infektionsphasen nach Symptombeginn hin.
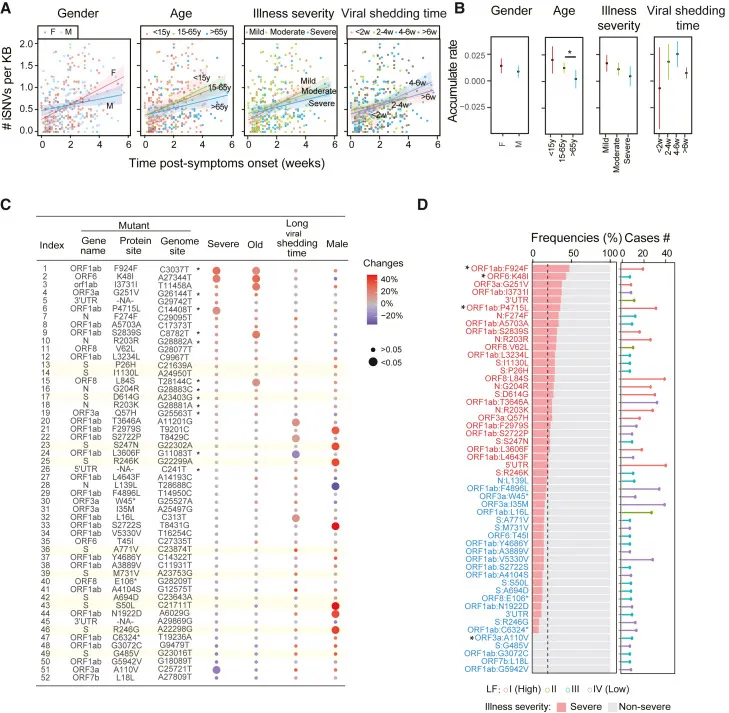 Verteilung von iSNVs in verschiedenen Patientengruppen
Verteilung von iSNVs in verschiedenen Patientengruppen
Um spezifische Mutationen weiter zu untersuchen, die von Wirtseffekten beeinflusst werden könnten, verglichen wir die Anteile von Individuen mit und ohne rezidivierende mutierte Stellen. Wir konstruierten eine Matrix von Individuen mit oder ohne iSNVs basierend auf 52 iSNV-Stellen, die von mehr als sechs Individuen aus einer Gruppe von 170 gemeinsam genutzt wurden. Diese Individuen wurden in vier unabhängige Klassen eingeteilt: Geschlecht, Alter, Krankheitschwere und virale Ausscheidungsdauer. Jede iSNV-Stelle wurde einem unabhängigen Fisher-Exakttest unterzogen. Individuen mit schwerer Krankheit, ältere Menschen, Personen mit langer viraler Ausscheidungsdauer und Männer zeigten vorzugsweise eine signifikante Anreicherung bei 4, 5, 4 bzw. 8 iSNVs im Vergleich zu Individuen mit leichter/moderater Krankheit, Kindern/mittelalten Personen, Personen mit kurzer viraler Ausscheidungsdauer (<6 Wochen) und Frauen. Diese iSNVs waren in ORF1ab, S, N, ORF6 und ORF8 verteilt. Unter den 52 rezidivierenden iSNV-Stellen identifizierten wir 27 Stellen, die vorzugsweise bei Individuen mit schwerer Krankheit auftraten, von denen 12 mit zuvor berichteten hfSNP-Stellen in der öffentlichen Datenbank 2019nCoVR übereinstimmten. Im Gegensatz dazu überschnitten sich die 25 iSNVs, die vorzugsweise bei Individuen mit moderater Krankheit auftraten, nicht mit hfSNP-Stellen. Diese Anreicherung von hfSNP-Stellen wurde in keiner anderen Kategorie beobachtet, die basierend auf Geschlecht, Alter und viraler Ausscheidungsdauer stratifiziert wurde, was auf einen nicht-stochastischen Prozess hindeutet.
Ungleichmäßige reinigende Selektionsprozesse von iSNVs zu SNPs
Um die Mutationen zu identifizieren, die von iSNVs zu SNPs fixiert wurden, verglichen wir die Genomstellen von iSNVs und SNPs in der 2019nCoVR-Datenbank. Von 7.037 iSNVs waren 15,59% der iSNVs bereits vor unserem Beobachtungszeitraum (Mai 2020) als SNPs identifiziert worden, und 11,28% der iSNVs wurden von Mai 2020 bis Dezember 2020 fixiert, während die verbleibenden iSNVs (73,13%) nicht als SNPs fixiert wurden. Nicht-synonyme iSNVs zeigten eine geringere Fixierungsrate im Vergleich zu synonymen iSNVs (20,92 % gegenüber 41,12 %, p < 0,001, Fisher-Exaktest). Diese Erkenntnis wird durch ein Modell gestützt, bei dem nicht-synonyme iSNVs aufgrund positiver Selektion oder unvollständiger reinigender Selektion in einem Individuum mit hoher Frequenz auftreten, aber aufgrund reinigender Selektion unwahrscheinlicher sind, sich in der Population zu fixieren. Als Nächstes führten wir einen Fisher-Exakttest durch, um den Anteil fixierter Mutationen in jedem Gen zu vergleichen, und S sowie ORF1ab zeigten signifikant niedrigere Fixierungsraten (21,04 % bzw. 20,56 %, p = 0,005 bzw. p < 0,001, Fisher-Exaktest) an nicht-synonymen und synonymen Stellen. Die Verhältnisse von nicht-synonym zu synonym von iSNVs in ORF1ab, N und S (ohne D614G) waren höher als die geschätzten für die identifizierten SNPs, was mit einer ungleichmäßigen reinigenden Selektion dieser Gene übereinstimmt. Mit fortschreitender Krankheit waren die iSNV-Fixierungsraten an nicht-synonymen und synonymen Stellen in der Population stabil, was auf einen Prozess ähnlicher reinigender Selektion im Zuge der Krankheit hindeutet. Wir beobachteten auch, dass Mutationen in iSNVs auftreten konnten, bevor sie als SNPs fixiert wurden. Zum Beispiel wurde die Akkumulation von C7051T-Allelen in unserer Studie vor Mai 2020 beobachtet, während der erste C7051T-SNP erst im Juni 2020 berichtet wurde. Obwohl Stichprobenverzerrungen auch unsere Beobachtung von SNPs in den frühen Stadien der Epidemie begrenzt haben könnten, könnten unsere Beobachtungen von Mutationen auf iSNV-Ebene unsere Nachweissensitivität für Mutationen erhöht haben, bevor sie fixiert wurden. Diese Ergebnisse deuten darauf hin, dass iSNVs eine ergänzende Ressource genetischer Informationen sind, um die Evolutionsgeschichte von SARS-CoV-2 zu beleuchten.
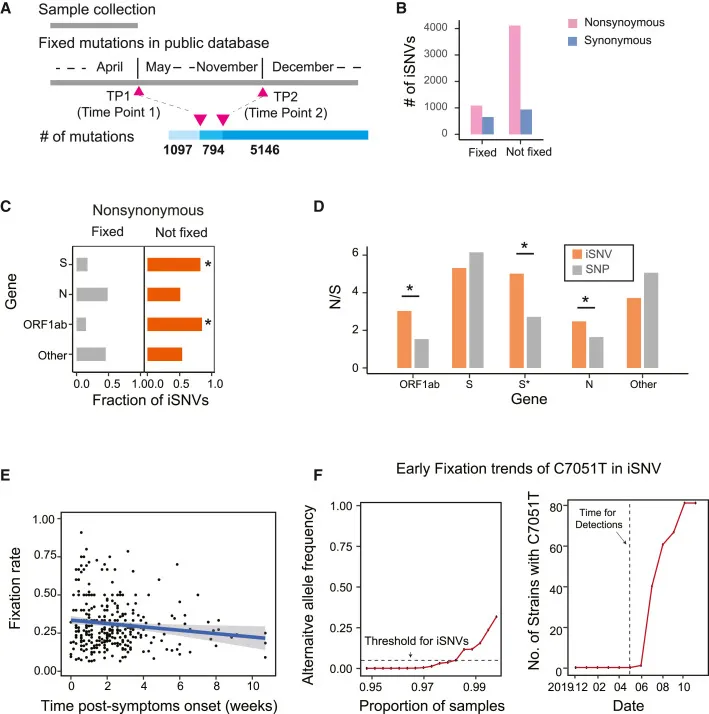 Biased Fixation von iSNVs in öffentlichen SNP-Datenbanken
Biased Fixation von iSNVs in öffentlichen SNP-Datenbanken
Molekulare Funktionen von S-Protein-Varianten vor reinigender Selektion
Das S-Protein treibt die Zellbindung und den Eintritt durch Rezeptoren an und fungiert als wichtiger Bestimmungsfaktor für Wirtsspektrum, Zelltyp, Gewebetropismus und Pathogenität von Coronaviren. Daher analysierten wir 21 nicht-synonyme Stellen von 606 iSNV-Stellen, die im kodierenden Bereich des S-Proteins identifiziert wurden und 20 Aminosäureänderungen verursachten: Neun wurden außerhalb der Rezeptor-Bindungsdomäne (RBD) bei mehr als sechs Individuen nachgewiesen, einschließlich der Substitution von zwei Aminosäureänderungen in drei verknüpften iSNVs. Elf befanden sich innerhalb der RBD oder an S1/S2-Spaltungsstellen bei mehr als zwei Individuen, einschließlich sieben iSNVs im Rezeptor-Bindungsmotiv (RBM). Da nur wenige dieser Mutationen in SARS-CoV-2 berichtet worden waren, verglichen wir die Mutationsstellen von sieben iSNVs im RBM von SARS-CoV-2 mit den Konsenssequenzen von SARS-CoV-2-ähnlichen Coronaviren bei anderen Tieren (Fledermäuse und Schuppentiere), um ihre potenziellen molekularen Funktionen zu erforschen. Alle diese Stellen waren heterogen, was darauf hindeutet, dass Mutationen an diesen Proteinpositionen keine zufälligen Mutationen sein könnten. Darüber hinaus hatten Individuen mit diesen Mutationen keine Kontaktgeschichte, mit Ausnahme von zwei Individuen mit der G485V-Mutation. Keine iSNVs traten bei Individuen zum ersten Zeitpunkt der Genomsequenzierung auf, und es gab keine Hinweise darauf, dass diese iSNVs im Genom verknüpft waren. Diese Ergebnisse deuten somit darauf hin, dass iSNVs in der RBD durch unabhängige virale Evolution generiert werden.
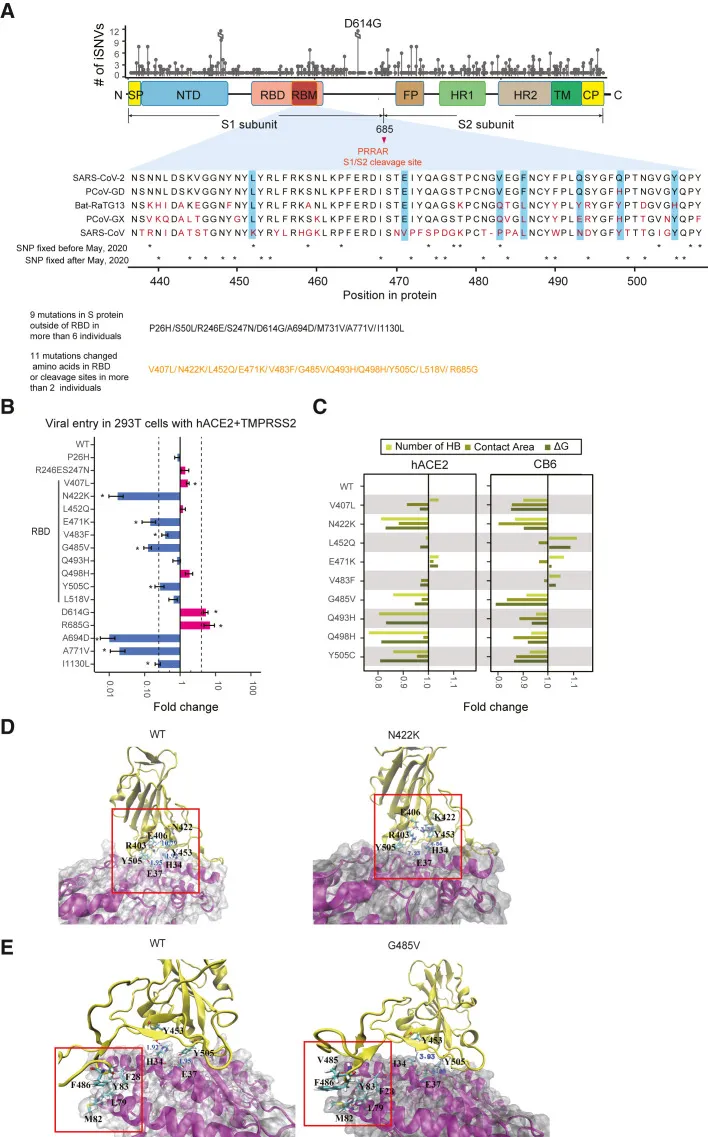 Genetische und molekulare Struktur-Analyse von iSNVs im S-Protein-Gen
Genetische und molekulare Struktur-Analyse von iSNVs im S-Protein-Gen
Um die Effekte dieser Mutationen auf molekularer Ebene zu beleuchten, verwendeten wir zunächst einen SARS-CoV-2-Pseudovirus-Infektionsassay in HEK293T-Zellen, um die virale Eintrittseffizienz von 20 der 22 S-Protein-Mutanten zu bewerten. Im Vergleich zum Referenzstamm zeigten 18 der 20 getesteten Mutanten eine verringerte (Veränderungsfaktor, <0,25; p < 0,05, t-Test) oder vergleichbare (Veränderungsfaktor, 0,25) virale Eintrittseffizienz; nur die Mutanten R685G und D614G zeigten eine ähnlich erhöhte virale Eintrittseffizienz. Da die L518V-Mutation weit von der Bindungsstelle zwischen dem S-Protein und hACE2/CB6 entfernt ist und vier Mutanten signifikant verringerte virale Eintrittseffizienz aufwiesen, wurden die anderen fünf RBD-Mutanten auf Empfindlichkeit gegenüber CB6 getestet. Wildtyp (WT) und D614G-Viren wurden als Kontrollen einbezogen. Mäßige Unterschiede zwischen diesen RBD-Varianten und dem Referenzstamm (≤4-fach) wurden in Bezug auf die Anfälligkeit für CB6 beobachtet. Einige Varianten zeigten sogar eine höhere Empfindlichkeit gegenüber CB6 als der Referenzstamm, was darauf hindeutet, dass CB6-Antikörper den Viruseintritt trotz RBD-Mutationen blockieren konnten.
Anschließend konzentrierten wir uns auf Mutationen innerhalb des RBM, indem wir die Bindung der entsprechenden Mutanten an menschliches Angiotensin-Converting-Enzym 2 (hACE2) und CB6-neutralisierende Antikörper mithilfe von molekularen Dynamiksimulationen simulierten. Die Simulation der L518V-Mutation wurde aus den beschriebenen Gründen nicht durchgeführt. Zum Vergleich simulierten wir auch die Bindung des WT-RBD an hACE2 und CB6. Die Cα-Wurzelmittelquadratabweichungs-Werte (RMSD) der Komplexe verschiedener mutierter RBDs, die an hACE2 binden, variierten in einem Bereich nahe dem Cα-RMSD-Wert des entsprechenden Komplexes mit dem WT-RBD, was darauf hindeutet, dass die neun Mutationen keine drastischen Konformationsänderungen hervorriefen.
Weitere Untersuchungen struktureller Merkmale ergaben, dass unterschiedliche Mutationen die Bindung des RBD an hACE2 auf verschiedene Weise beeinflussen. Beispielsweise wurde der Rückstand 422 zu einem positiv geladenen K mutiert, was zu einem viel stärkeren Wasserstoffbrücken (Salzbrücke) zwischen K422 und E406 führte. Darüber hinaus verursachten starke Abstoßungen zwischen K422 und R403 sowie Anziehungen zwischen K422 und Y453 den Bruch von Wasserstoffbrücken zwischen Y505 und E37 sowie zwischen Y453 und H34. Infolgedessen wurden die Anzahl der Wasserstoffbrücken und die Kontaktfläche zwischen den mutierten RBDs und hACE2 reduziert. Im Hinblick auf die G485V-Mutation hat V eine relativ voluminöse Seitenkette im Vergleich zu G; um diese voluminöse Seitenkette neu zu positionieren, führte die G485V-Mutation zum Ausstoßen von F486 aus der hydrophoben Tasche, die von Rückständen gebildet wird. Infolgedessen wurden die Kontaktfläche und die Anzahl der Wasserstoffbrücken zwischen dem mutierten RBD und hACE2 reduziert. Wichtig ist, dass die Wasserstoffbrücke zwischen Y505 des S-Proteins und E37 von hACE2, die für die Bindung wichtig ist, verloren ging. Als mögliche Folge war die Bindung von Mutanten N422K/G485V an hACE2 geschwächt. Berechnungen der Bindungsfreie Energie mit der Molekularen Mechanik Energien kombiniert mit der Verallgemeinerten Born- und Oberflächenflächen-Kontinuumslösung (MM/GBSA)-Methode zeigten ebenfalls eine geschwächte Bindung von N422K/G485 an hACE2.
Änderungen der Wasserstoffbrücken und der Kontaktfläche wurden auch für andere Mutanten untersucht. Die meisten Mutanten mit iSNVs zeigten eine verringerte virale Eintrittsfähigkeit aufgrund geringerer Wasserstoffbrücken und/oder reduzierter Kontaktfläche zwischen mutierten RBDs und hACE2. Wir analysierten auch Simulationsergebnisse für Komplexe zwischen mutierten RBDs und dem CB6-Antikörper. Die meisten Mutanten zeigten einen stark reduzierten Kontaktbereich im Vergleich zur WT-Sequenz. Eine verringerte Bindungsaffinität wurde aufgrund von weniger Wasserstoffbrücken beobachtet, was mit einer höheren Bindungsfreien Energie in den MM/GBSA-Berechnungen übereinstimmt. Daher deuteten die Beobachtung von Mutationen in den pseudoviralen Infektionsassays und die Berechnung ihrer Wechselwirkungsenergien auf eine geschwächte virale Infektion für die meisten im S-Protein identifizierten iSNVs hin.
Diskussion
Die anhaltende SARS-CoV-2-Pandemie ist von weltweiter Bedeutung. SARS-CoV-2-Mutationen entstehen natürlich, während das Virus repliziert, und die resultierenden SNPs können Selektion und Übertragung beeinflussen. Innerhalb eines Jahres nach Bestätigung des ersten COVID-19-Falls wurden Tausende von Mutationen, die SNPs beinhalten, identifiziert, von denen nur eine sehr kleine Minderheit Veränderungen in der Infektiosität und Immun-Evasion von SARS-CoV-2 verursachte. Mutationen im SARS-CoV-2-Genom können verwendet werden, um Anzeichen von Selektion zu erkennen, die sich während der Virusentwicklung ansammeln. In der vorliegenden Studie haben wir gezeigt, dass die intra-host-Variation bei SARS-CoV-2 in einer Reihe von mehr als 400 klinischen Proben nicht zufällig über das Genom verteilt war, was auf eine Rolle der Selektion hindeutet. Im Vergleich zu synonymen Mutationen waren nicht-synonyme Mutationen in iSNVs überrepräsentiert, aber in SNPs (Populationsniveau) unterrepräsentiert. Molekulare Funktionsanalysen der Auswirkungen von Mutationen im S-Protein sowie Korrelationsanalysen zwischen Mutationen und klinischen Merkmalen deuteten darauf hin, dass Mutationen, die die Krankheitschwere erhöhen oder die Immun-Evasion begünstigen, selten sind, was die geringe Anzahl nicht-synonymer Mutationen in Populationen erklären könnte.
Unser Sequenzierungsansatz lieferte Beweise für eine zweistufige “Step Fitness”-Selektion für intra-host-Varianten bei SARS-CoV-2. Die erste Stufe der Selektion tritt nach der Generierung zufälliger Mutationen auf, und positive Selektion (z.B. individuell abgeleitete “Step Fitness”-Selektion) vermittelt die Akkumulation nicht-synonymer iSNVs, was darauf hindeutet, dass die genetische Diversität mit fortschreitender COVID-19 zunimmt. Positive Selektion führt zu zwei Merkmalen von iSNVs. Erstens eine Zunahme der genetischen Diversität mit fortschreitender COVID-19. RNA-Editing und Wirtsimmunität können diesen Prozess beeinflussen. Unsere Ergebnisse zeigen, dass die Rate der iSNV-Akkumulation bei mittelalten Individuen signifikant höher ist als bei älteren. Es gibt Hinweise darauf, dass ältere Menschen unter höherem Immun-Druck stehen, was mit der Akkumulationsrate von iSNVs verbunden sein kann. Das zweite Merkmal ist eine ungleichmäßige Verteilung von iSNVs zwischen Individuen und Genomen. Die rezidivierenden iSNVs bei verschiedenen Individuen implizieren auch, dass diese Stellen unter Selektionsdruck standen, einschließlich positiver Selektion und unvollständiger reinigender Selektion. Die Tendenz zu mehr iSNVs mit zunehmender Allelfrequenz bei einem Individuum deutet darauf hin, dass diese Stellen unter positiver Selektion standen. Darüber hinaus akkumulieren sich diese Mutationen vorzugsweise an nicht-synonymen Mutationsstellen, was wichtige Merkmale des Virus beeinflussen könnte, einschließlich Infektiosität, Virulenz und Immunogenität. Mehrere andere Studien an anderen Viren, wie dem Ebola-Virus, Lassa-Virus und Influenza-Virus, unterstützen die Existenz intra-host positiver Selektion. Hohe Raten der Mutationsakkumulation über kurze Zeiträume bei SARS-CoV-2 wurden in früheren Studien an immundefizienten oder immunsupprimierten Patienten mit chronischer SARS-CoV-2-Infektion berichtet. Darüber hinaus identifizierten mehrere neuere Veröffentlichungen zum SARS-CoV-2-Genom Signale positiver Selektion und Konservierung innerhalb des Gens, das für das S-Protein kodiert.
Die zweite Stufe im Selektionsprozess ist die reinigende Selektion, die für die Reduzierung der Anzahl nicht-synonymer Mutationen beim Übergang von iSNVs zu SNPs verantwortlich ist. Dies hat drei Implikationen: (1) Frequenzabhängige Wechselwirkungen und klonale Interferenz können wichtige Kräfte sein, die den Selektionsprozess antreiben. (2) Ob das Nachkommen-Virus eine produktive Infektion etablieren kann, kann die Verbreitung von Mutationen in einer Population beeinflussen. (3) Der Übertragungsengpass ist der bestimmende Faktor für produktive Infektionen zwischen Wirten und zwischen Zellen in einem einzelnen Individuum. Da die in Peking beprobten Individuen meist importierte und/oder sporadische Fälle waren, war es schwierig, die reinigende Selektion von iSNVs zu SNPs in der vorliegenden Studie zu messen. Diese aktuellen Erkenntnisse liefern Einblicke in das Auftreten von Mutationen weltweit sowie eine Richtung für Bemühungen zur Bekämpfung der globalen COVID-19-Pandemie.
Während sich nicht-synonyme Mutationen bei Individuen akkumulierten, beobachteten wir auch 760 nicht-synonyme Mutationen im S-Protein. Obwohl das In-vitro-Experiment und die molekulare Dynamiksimulation bewiesen, dass die virale Eintrittseffizienz von hochfrequenten iSNV-Mutationen tendenziell geschwächt ist, bleibt unklar, ob diese geschwächten Viren über lange Zeiträume bei Personen mit Long-COVID-19-Infektion persistieren. Mutationen in Populationen sind jedoch unter den aktuellen Umständen zu erwarten. Änderungen der Umstände, wie die weit verbreitete Einführung von Antikörpertherapien, werden den Selektionsdruck auf jede Mutation verändern. Neu auftretende Mutationen wie N501Y, E484K und L452R zeigen ihr Potenzial zur Immun-Evasion gegenüber monoklonalen Antikörpern und Impfstoffen. Starker Selektionsdruck durch Antikörper und Impfstoffe könnte zu einer schnellen Umgestaltung der Virusgenetik durch direkte Selektion oder genetische Drift führen. Daher werden mit größerer Wahrscheinlichkeit weitere Mutationen von SARS-CoV-2 als iSNVs beobachtet, zusammen mit der R685G-Mutation an Spaltungsstellen, die die virale Eintrittseffizienz erhöht, zumindest in einigen Zelltypen. Diese Mutationen könnten die Persistenz von SARS-CoV-2 in mehreren Organen verstärken, obwohl solche SNPs noch nicht in öffentlichen Datenbanken identifiziert wurden.
Angesichts der Dringlichkeit der Impfstoffentwicklung und Behandlungsstrategien ist es möglicherweise nicht ratsam, auf die Fixierung von Mutationen in Populationen zu warten, bevor Anstrengungen unternommen werden, um deren Auswirkungen nachzuweisen. Wichtig ist, dass frühes Wissen über potenzielle Evolutionen die Impfstoffentwicklung leiten könnte. Zusammenhänge zwischen aufkommenden Mutationen und der Krankheitschwere sowie Behandlungen sollten sorgfältig berücksichtigt werden. Mehr genomische Daten auf intra-host-Ebene sollten gesammelt werden, um die Untersuchung der genetischen Selektion auf Genomebene zu ermöglichen, einschließlich der potenziellen Auswirkungen auf die Krankheitschwere, klinische Ergebnisse und die Anfälligkeit verschiedener Populationen.
Einschränkungen der Studie
Unsere Studie beschreibt die dynamische Veränderung von iSNVs bei Individuen. Aufgrund der begrenzten Anzahl von Individuen konnten wir jedoch keine signifikanten Unterschiede in verschiedenen Gruppen feststellen. Die Beziehung zwischen Immun-Druck und iSNV-Dynamik wird eine interessante und wichtige zukünftige Studie sein.